

Lipotoxic hepatocyte derived LIMA1 enriched small extracellular vesicles promote hepatic stellate cells activation via inhibiting mitophagy
- PMID: 38822260
- PMCID: PMC11140962
- DOI: 10.1186/s11658-024-00596-4
Lipotoxic hepatocyte derived LIMA1 enriched small extracellular vesicles promote hepatic stellate cells activation via inhibiting mitophagy
Abstract
Background: Hepatic stellate cells (HSCs) play a crucial role in the development of fibrosis in non-alcoholic fatty liver disease (NAFLD). Small extracellular vesicles (sEV) act as mediators for intercellular information transfer, delivering various fibrotic factors that impact the function of HSCs in liver fibrosis. In this study, we investigated the role of lipotoxic hepatocyte derived sEV (LTH-sEV) in HSCs activation and its intrinsic mechanisms.
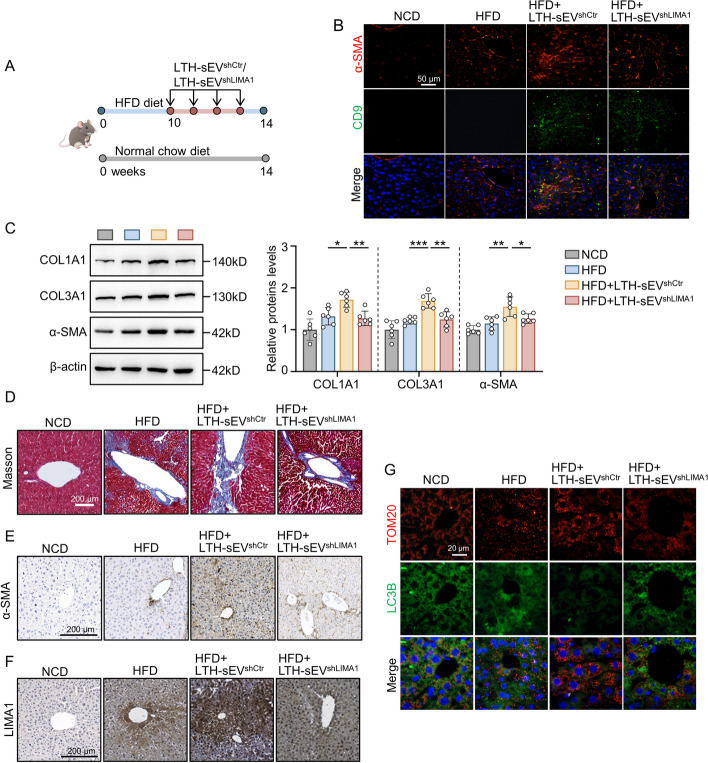
Methods: High-fat diet (HFD) mice model was constructed to confirm the expression of LIMA1. The relationship between LIMA1-enriched LTH-sEV and LX2 activation was evaluated by measurement of fibrotic markers and related genes. Levels of mitophagy were detected using mt-keima lentivirus. The interaction between LIMA1 and PINK1 was discovered through database prediction and molecular docking. Finally, sEV was injected to investigate whether LIMA1 can accelerate HFD induced liver fibrosis in mice.
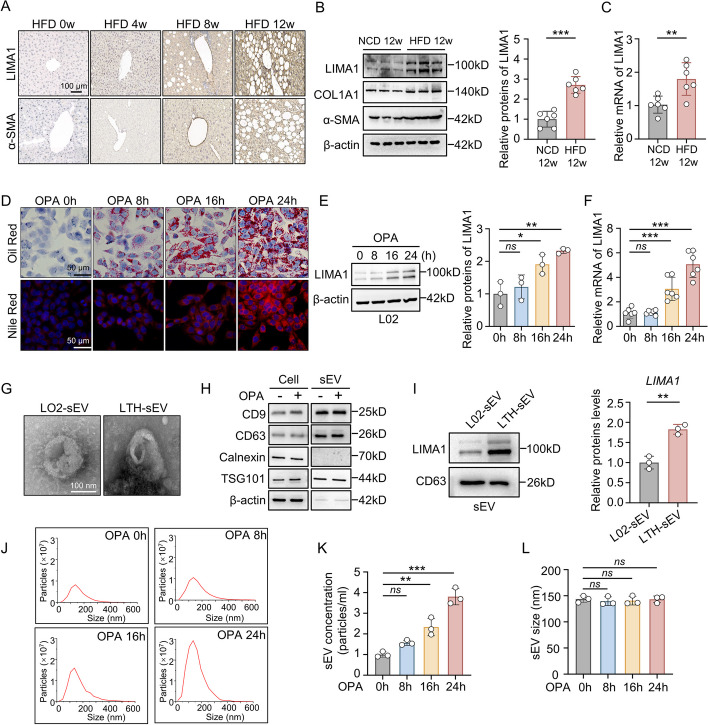
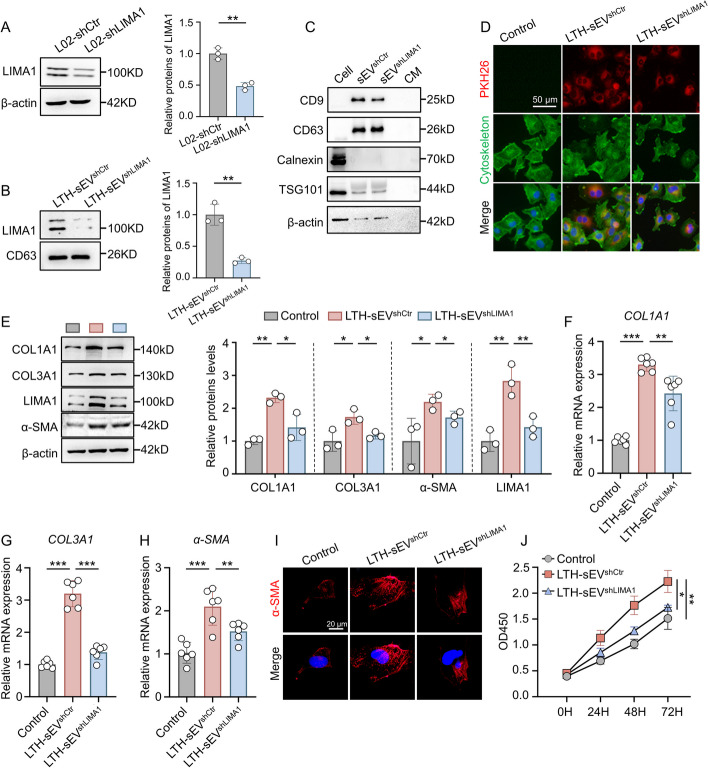
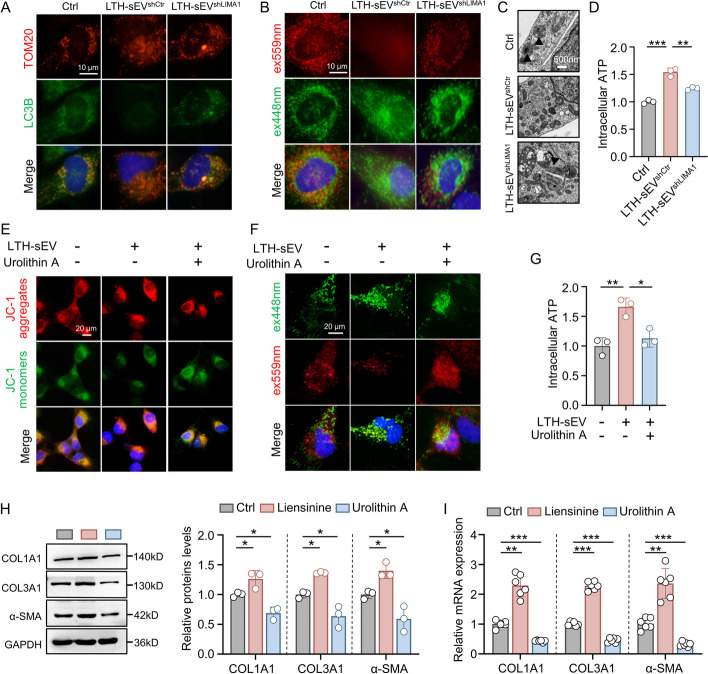
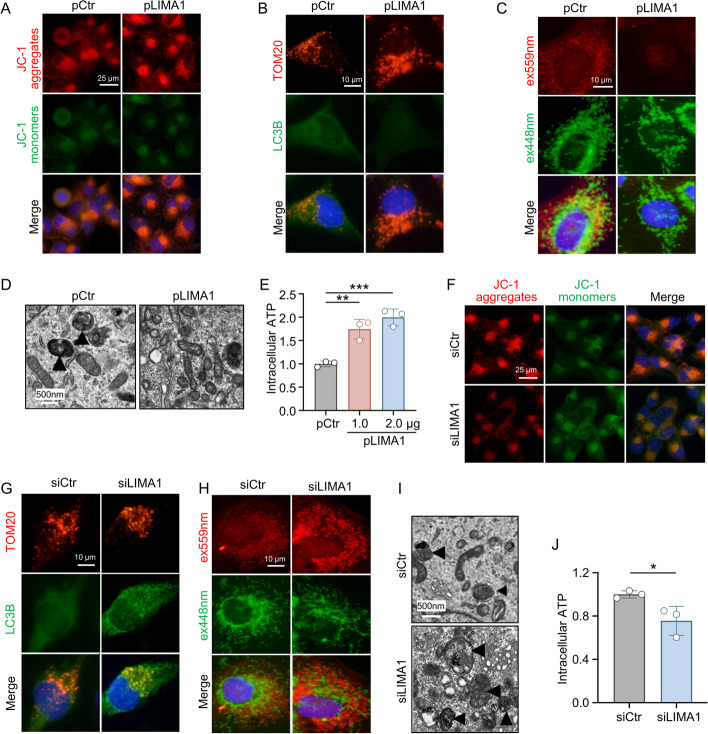
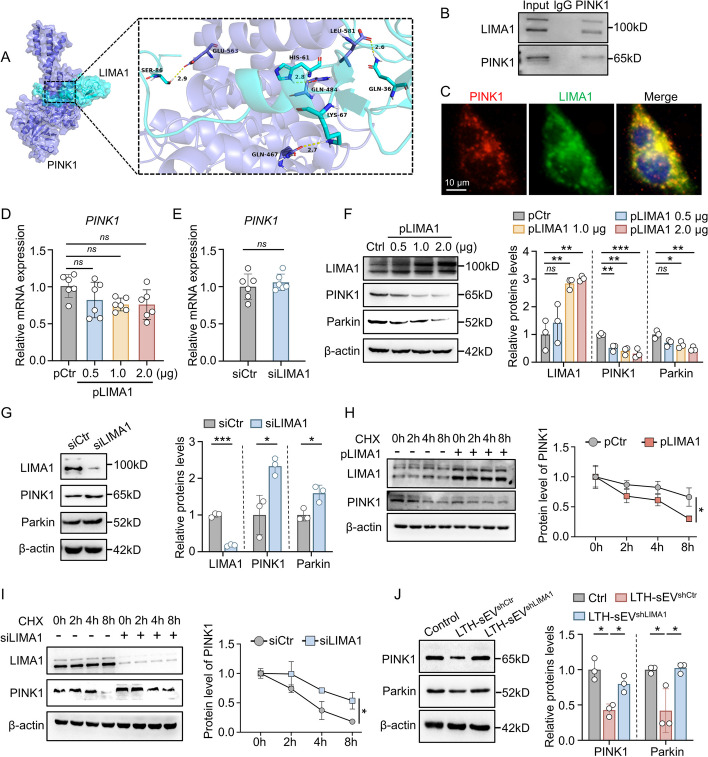
Results: LIMA1 expression was upregulated in lipotoxic hepatocytes and was found to be positively associated with the expression of the HSCs activation marker α-SMA. Lipotoxicity induced by OPA led to an increase in both the level of LIMA1 protein in LTH-sEV and the release of LTH-sEV. When HSCs were treated with LTH-sEV, LIMA1 was observed to hinder LX2 mitophagy while facilitating LX2 activation. Further investigation revealed that LIMA1 derived from LTH-sEV may inhibit PINK1-Parkin-mediated mitophagy, consequently promoting HSCs activation. Knocking down LIMA1 significantly attenuates the inhibitory effects of LTH-sEV on mitophagy and the promotion of HSCs activation.
Conclusions: Lipotoxic hepatocyte-derived LIMA1-enriched sEVs play a crucial role in promoting HSCs activation in NAFLD-related liver fibrosis by negatively regulating PINK1 mediated mitophagy. These findings provide new insights into the pathological mechanisms involved in the development of fibrosis in NAFLD.
Keywords: Hepatic stellate cells; LIMA1; Mitophagy; Nonalcoholic fatty liver disease; Small extracellular vesicles.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing or financial interest.
Figures


Similar articles
-
Aflatoxin B1-exposed hepatocyte-derived extracellular vesicles: Initiating hepatic stellate cell-mediated liver fibrosis through a p53-Parkin-dependent mitophagy pathway.Ecotoxicol Environ Saf. 2024 Jun 1;277:116363. doi: 10.1016/j.ecoenv.2024.116363. Epub 2024 Apr 25. Ecotoxicol Environ Saf. 2024. PMID: 38663190
-
CircRNA608-microRNA222-PINK1 axis regulates the mitophagy of hepatic stellate cells in NASH related fibrosis.Biochem Biophys Res Commun. 2022 Jun 25;610:35-42. doi: 10.1016/j.bbrc.2022.04.008. Epub 2022 Apr 5. Biochem Biophys Res Commun. 2022. PMID: 35436629
-
RNF31 alleviates liver steatosis by promoting p53/BNIP3-related mitophagy in hepatocytes.Free Radic Biol Med. 2024 Jul;219:163-179. doi: 10.1016/j.freeradbiomed.2024.04.214. Epub 2024 Apr 13. Free Radic Biol Med. 2024. PMID: 38615890
-
Extracellular Vesicles in the Development of the Non-Alcoholic Fatty Liver Disease: An Update.Biomolecules. 2020 Oct 30;10(11):1494. doi: 10.3390/biom10111494. Biomolecules. 2020. PMID: 33143043 Free PMC article. Review.
-
Role of G Protein-Coupled Receptors in Hepatic Stellate Cells and Approaches to Anti-Fibrotic Treatment of Non-Alcoholic Fatty Liver Disease.Front Endocrinol (Lausanne). 2021 Dec 6;12:773432. doi: 10.3389/fendo.2021.773432. eCollection 2021. Front Endocrinol (Lausanne). 2021. PMID: 34938271 Free PMC article. Review.
Cited by
-
From Genetic Findings to new Intestinal Molecular Targets in Lipid Metabolism.Curr Atheroscler Rep. 2025 Jan 11;27(1):26. doi: 10.1007/s11883-024-01264-w. Curr Atheroscler Rep. 2025. PMID: 39798054 Review.
-
Pyrimethamine Inhibits Human Ovarian Cancer by Triggering Lethal Mitophagy via Activating the p38/JNK/ERK Pathway.Oncol Res. 2025 Aug 28;33(9):2421-2434. doi: 10.32604/or.2025.063724. eCollection 2025. Oncol Res. 2025. PMID: 40918465 Free PMC article.
-
Extracellular Vesicles as Biomarkers in Chronic Hepatobiliary Diseases: An Overview of Their Interplay.Int J Mol Sci. 2025 Jun 30;26(13):6333. doi: 10.3390/ijms26136333. Int J Mol Sci. 2025. PMID: 40650114 Free PMC article. Review.
-
Communication initiated by hepatocytes: The driver of HSC activation and liver fibrosis.Hepatol Commun. 2025 Jul 14;9(8):e0753. doi: 10.1097/HC9.0000000000000753. eCollection 2025 Aug 1. Hepatol Commun. 2025. PMID: 40658809 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
