

Knockout mice with pituitary malformations help identify human cases of hypopituitarism
- PMID: 38822427
- PMCID: PMC11140907
- DOI: 10.1186/s13073-024-01347-y
Knockout mice with pituitary malformations help identify human cases of hypopituitarism
Abstract
Background: Congenital hypopituitarism (CH) and its associated syndromes, septo-optic dysplasia (SOD) and holoprosencephaly (HPE), are midline defects that cause significant morbidity for affected people. Variants in 67 genes are associated with CH, but a vast majority of CH cases lack a genetic diagnosis. Whole exome and whole genome sequencing of CH patients identifies sequence variants in genes known to cause CH, and in new candidate genes, but many of these are variants of uncertain significance (VUS).
Methods: The International Mouse Phenotyping Consortium (IMPC) is an effort to establish gene function by knocking-out all genes in the mouse genome and generating corresponding phenotype data. We used mouse embryonic imaging data generated by the Deciphering Mechanisms of Developmental Disorders (DMDD) project to screen 209 embryonic lethal and sub-viable knockout mouse lines for pituitary malformations.
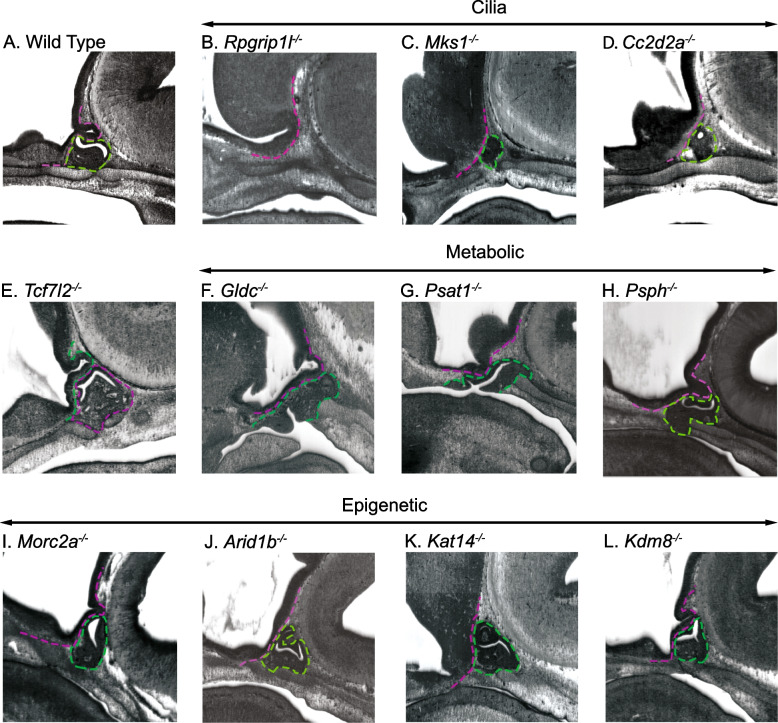
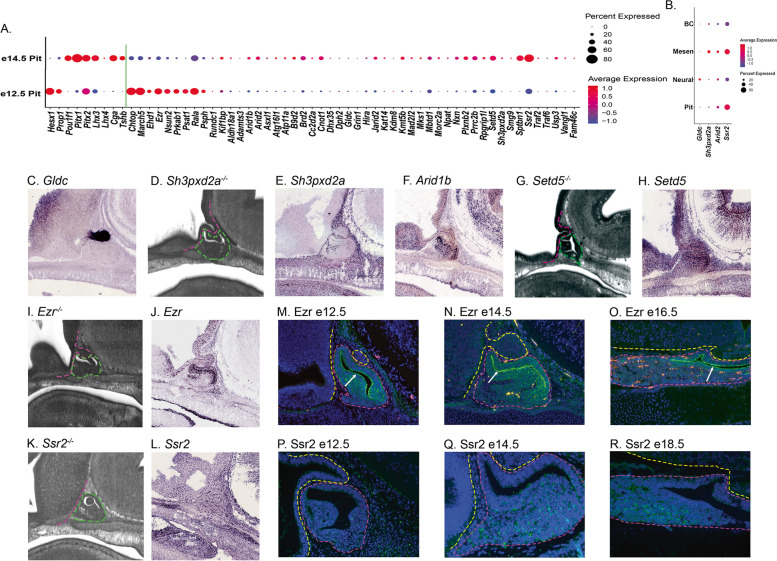
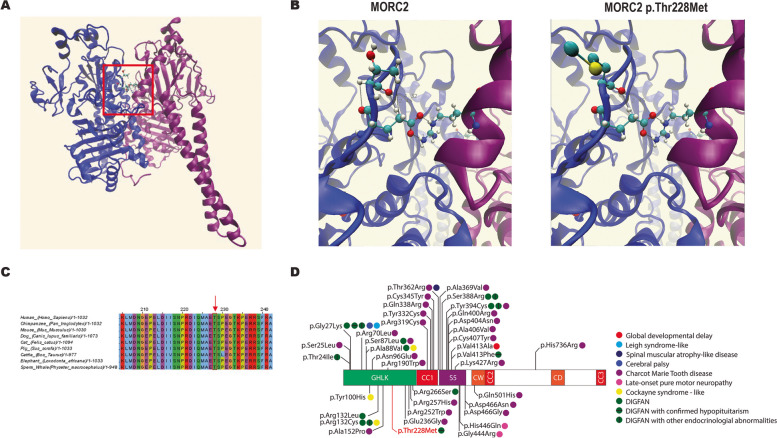
Results: Of the 209 knockout mouse lines, we identified 51 that have embryonic pituitary malformations. These genes not only represent new candidates for CH, but also reveal new molecular pathways not previously associated with pituitary organogenesis. We used this list of candidate genes to mine whole exome sequencing data of a cohort of patients with CH, and we identified variants in two unrelated cases for two genes, MORC2 and SETD5, with CH and other syndromic features.
Conclusions: The screening and analysis of IMPC phenotyping data provide proof-of-principle that recessive lethal mouse mutants generated by the knockout mouse project are an excellent source of candidate genes for congenital hypopituitarism in children.
Keywords: MORC2; SETD5; Cleft palate; Development; Growth hormone; Holoprosencephaly; Hypothalamus; International Mouse Phenotyping Consortium (IMPC); Pituitary; Septo-optic dysplasia.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
-
- Blum WF, Klammt J, Amselem S, Pfäffle HM, Legendre M, Sobrier ML, et al. Screening a large pediatric cohort with GH deficiency for mutations in genes regulating pituitary development and GH secretion: Frequencies, phenotypes and growth outcomes. EBioMedicine. 2018;36:390–400. doi: 10.1016/j.ebiom.2018.09.026. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- 2021162/PICT
- R01 HD097096/HD/NICHD NIH HHS/United States
- R03DE031037/DE/NIDCR NIH HHS/United States
- R15HD107430/National Institute of Child Health and Human Development
- End the Diagnostic Odyssey/3Billion
- R03 DE031037/DE/NIDCR NIH HHS/United States
- R01HD108156/National Institute of Child Health and Human Development
- R03AG072221/Division of Intramural Research, National Institute of Allergy and Infectious Diseases
- R01HD097096/National Institute of Child Health and Human Development
- R03 AG072221/AG/NIA NIH HHS/United States
- R01 HD108156/HD/NICHD NIH HHS/United States
- R15 HD107430/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
