

Inherited C-terminal TREX1 variants disrupt homology-directed repair to cause senescence and DNA damage phenotypes in Drosophila, mice, and humans
- PMID: 38824133
- PMCID: PMC11144269
- DOI: 10.1038/s41467-024-49066-7
Inherited C-terminal TREX1 variants disrupt homology-directed repair to cause senescence and DNA damage phenotypes in Drosophila, mice, and humans
Abstract
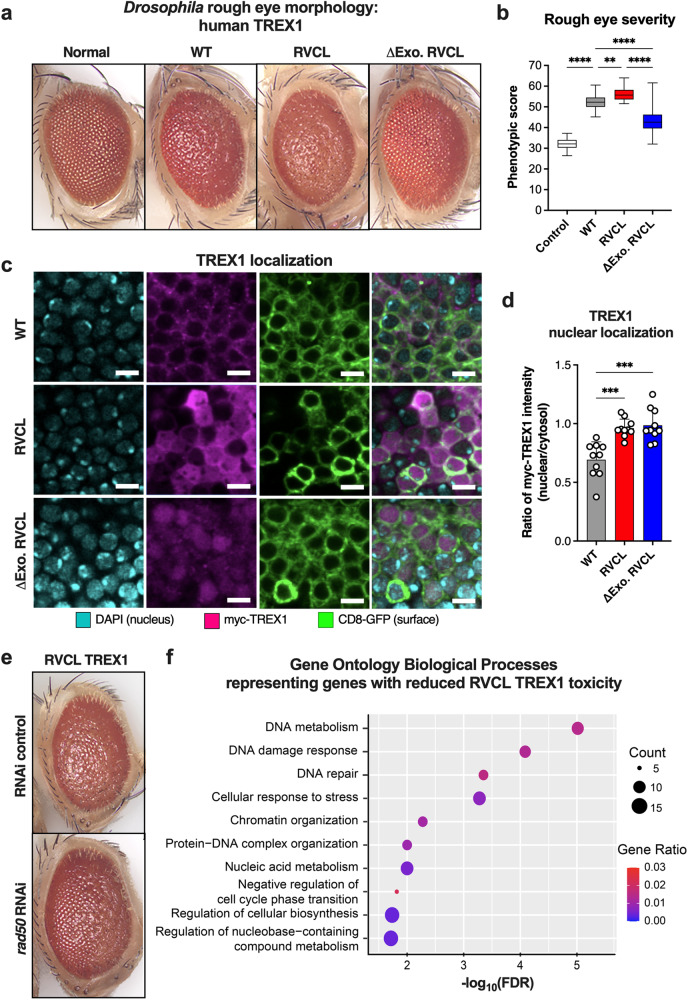
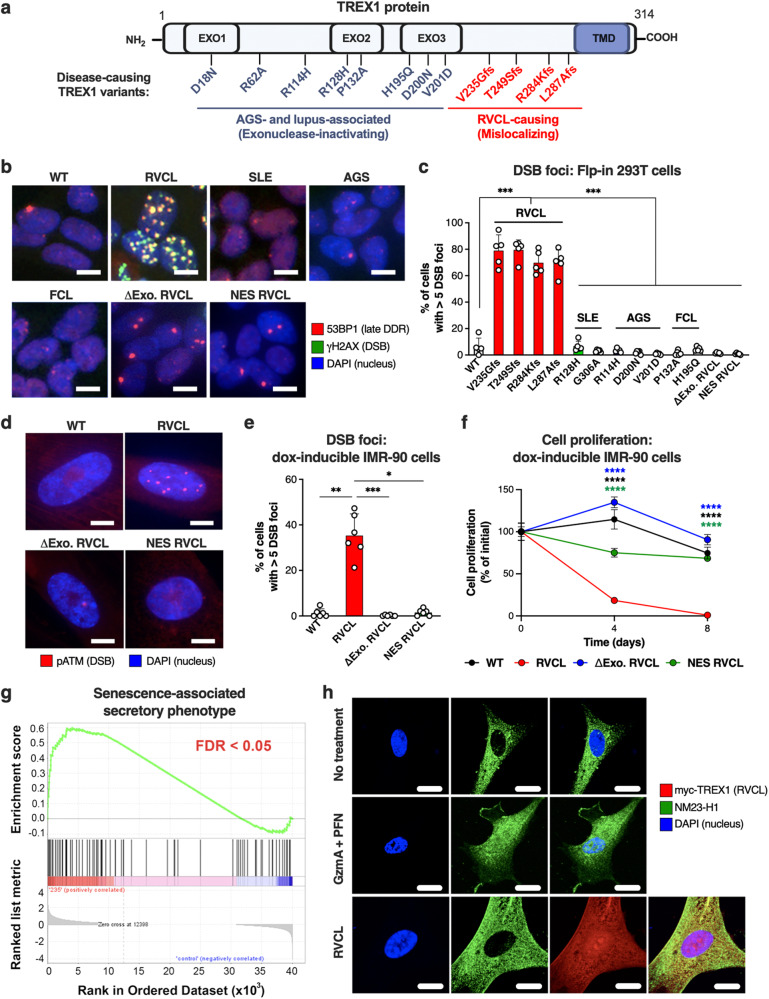
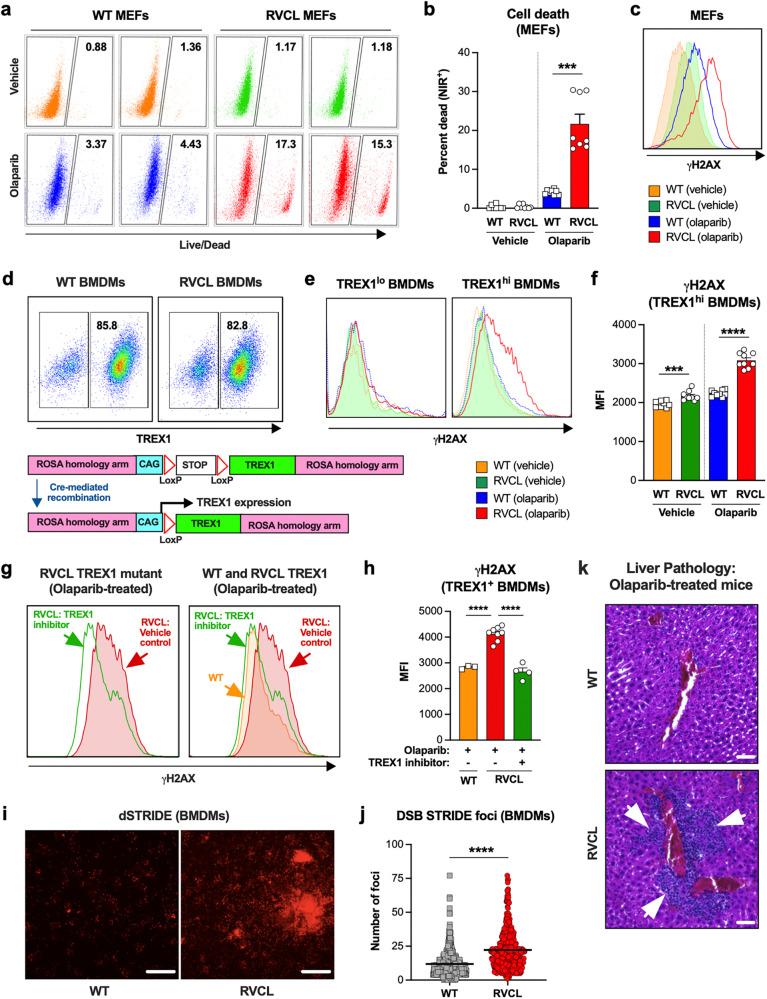
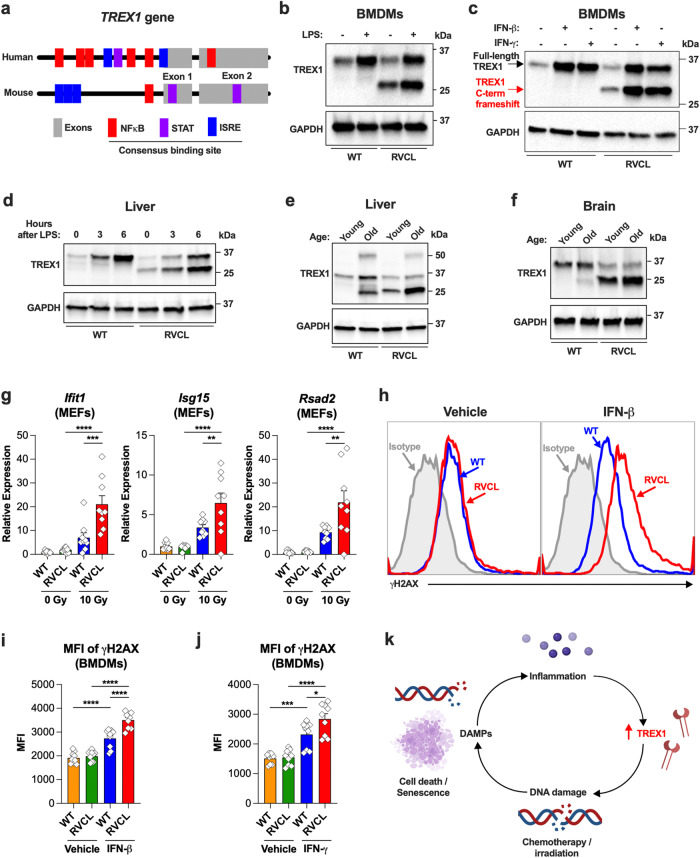
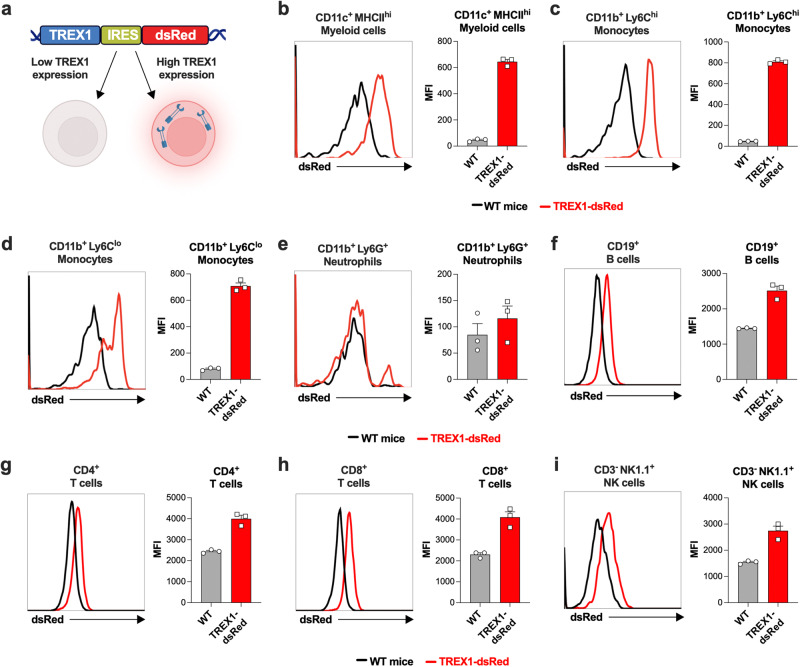
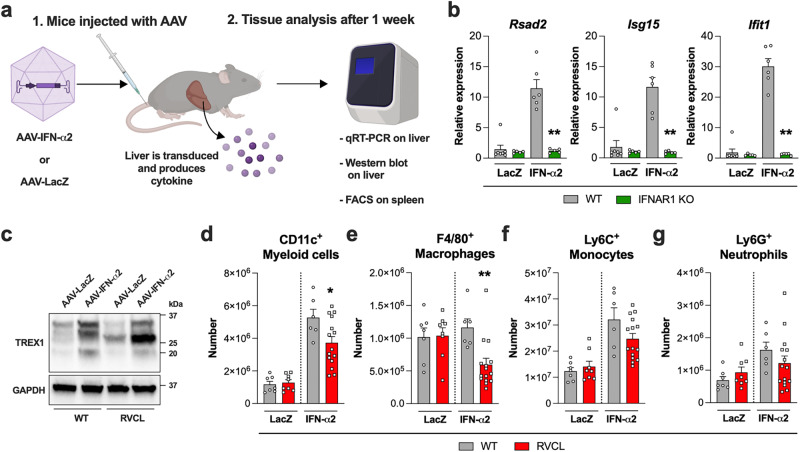
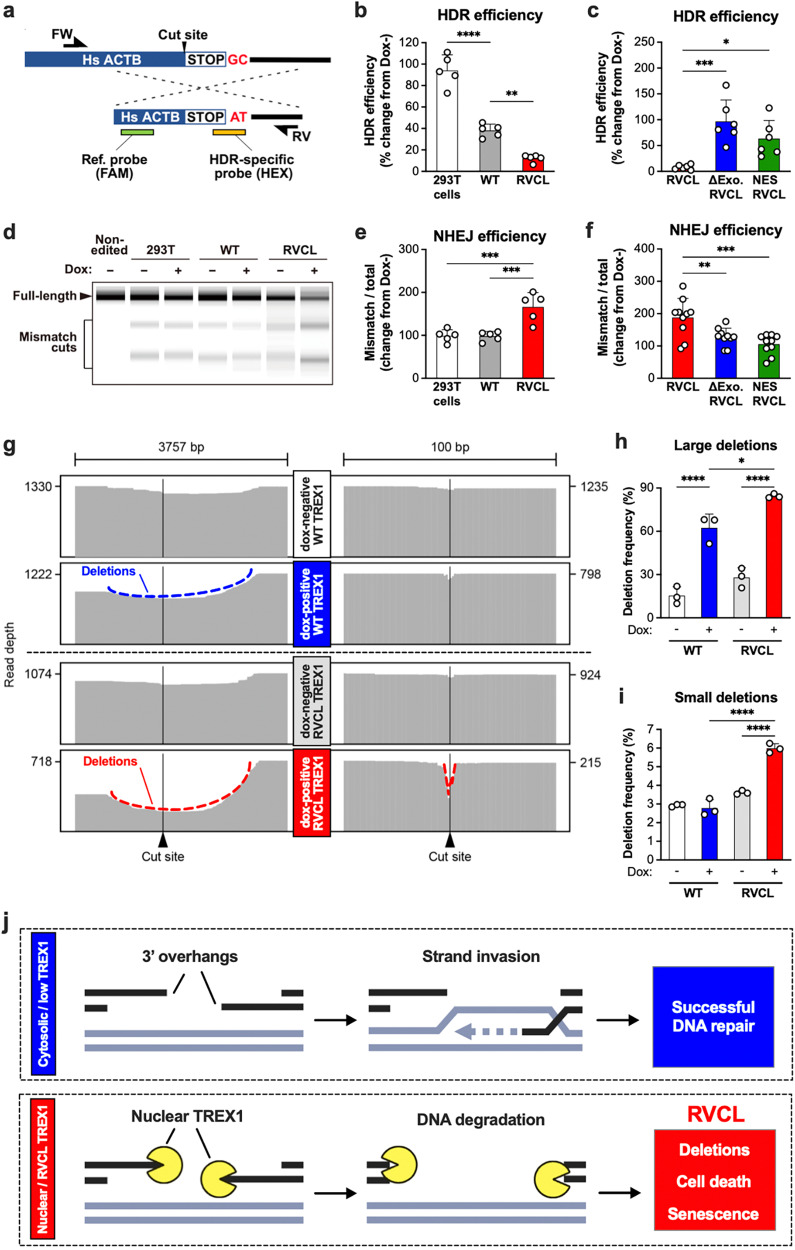
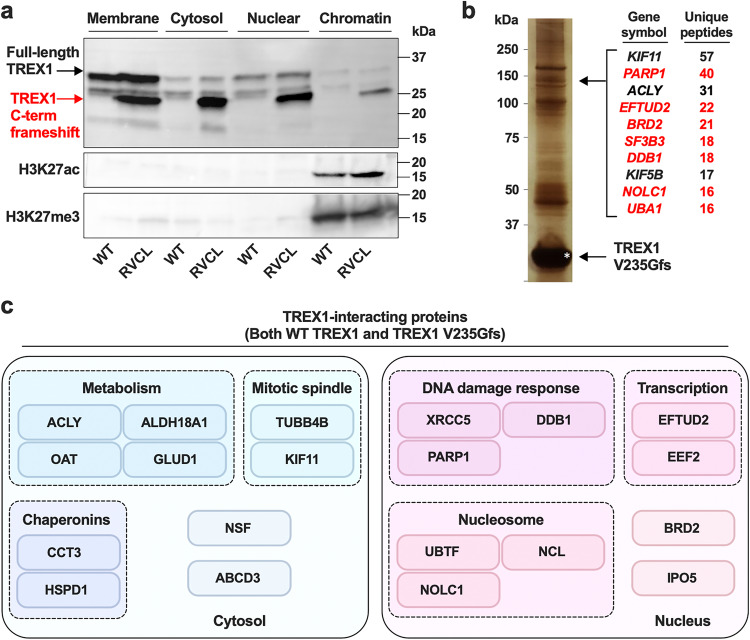
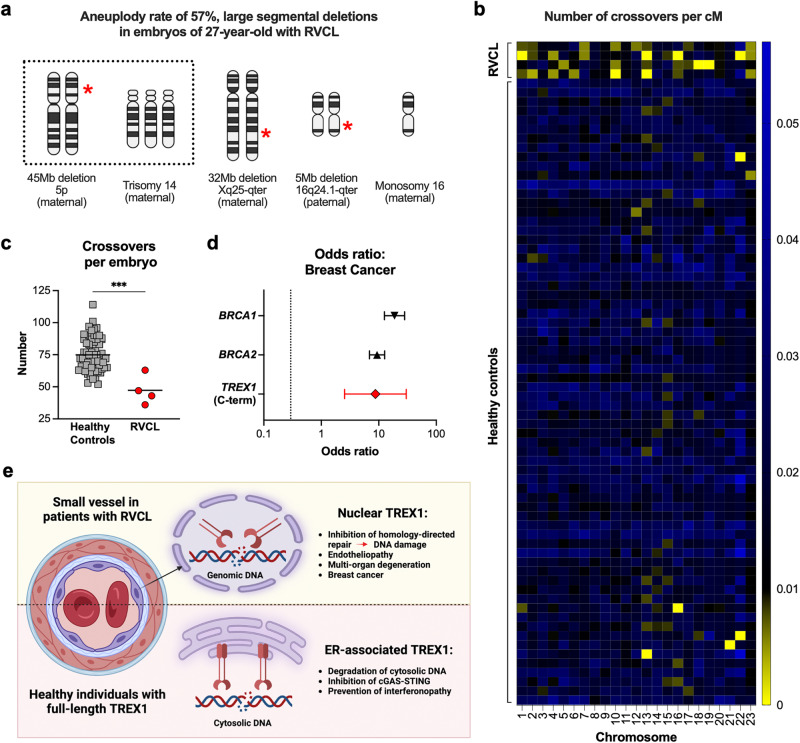
Age-related microangiopathy, also known as small vessel disease (SVD), causes damage to the brain, retina, liver, and kidney. Based on the DNA damage theory of aging, we reasoned that genomic instability may underlie an SVD caused by dominant C-terminal variants in TREX1, the most abundant 3'-5' DNA exonuclease in mammals. C-terminal TREX1 variants cause an adult-onset SVD known as retinal vasculopathy with cerebral leukoencephalopathy (RVCL or RVCL-S). In RVCL, an aberrant, C-terminally truncated TREX1 mislocalizes to the nucleus due to deletion of its ER-anchoring domain. Since RVCL pathology mimics that of radiation injury, we reasoned that nuclear TREX1 would cause DNA damage. Here, we show that RVCL-associated TREX1 variants trigger DNA damage in humans, mice, and Drosophila, and that cells expressing RVCL mutant TREX1 are more vulnerable to DNA damage induced by chemotherapy and cytokines that up-regulate TREX1, leading to depletion of TREX1-high cells in RVCL mice. RVCL-associated TREX1 mutants inhibit homology-directed repair (HDR), causing DNA deletions and vulnerablility to PARP inhibitors. In women with RVCL, we observe early-onset breast cancer, similar to patients with BRCA1/2 variants. Our results provide a mechanistic basis linking aberrant TREX1 activity to the DNA damage theory of aging, premature senescence, and microvascular disease.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Supplementary concepts
Grants and funding
- K08AR070918/U.S. Department of Health & Human Services | NIH | National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS)
- 22ek0109424h0003/Japan Agency for Medical Research and Development (AMED)
- 22H02981/MEXT | Japan Society for the Promotion of Science (JSPS)
- R01NS131480/U.S. Department of Health & Human Services | NIH | National Institute of Neurological Disorders and Stroke (NINDS)
- 18K07522/MEXT | Japan Society for the Promotion of Science (JSPS)
- R01AI143982/U.S. Department of Health & Human Services | NIH | National Institute of Allergy and Infectious Diseases (NIAID)
- R01 AI143982/AI/NIAID NIH HHS/United States
- R01 NS131480/NS/NINDS NIH HHS/United States
- K08 AR070918/AR/NIAMS NIH HHS/United States
- 22H00466/MEXT | Japan Society for the Promotion of Science (JSPS)
- P01 CA265794/CA/NCI NIH HHS/United States
- T32 GM007170/GM/NIGMS NIH HHS/United States
- R01 GM101149/GM/NIGMS NIH HHS/United States
- JP21FC1007/Osaka Medical Research Foundation for Intractable Diseases
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
