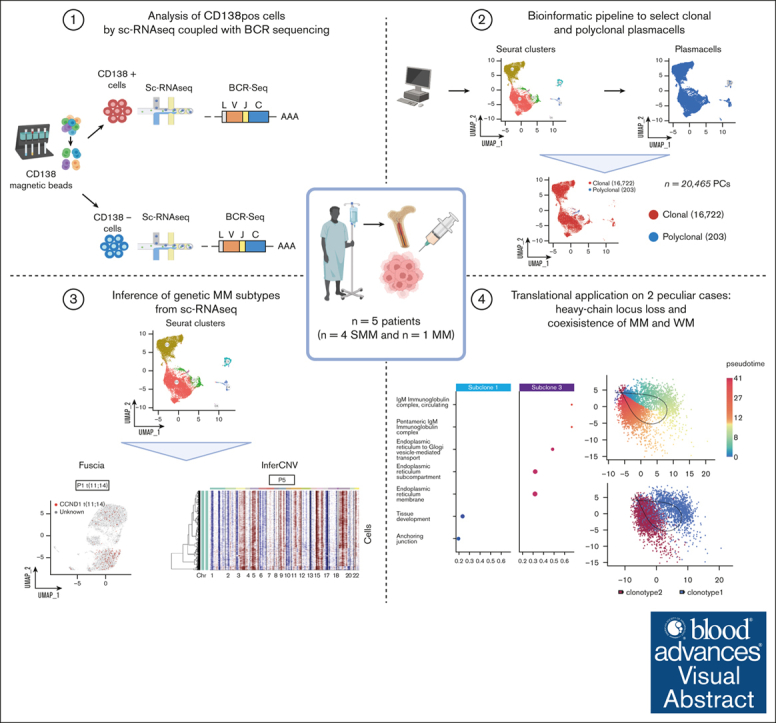
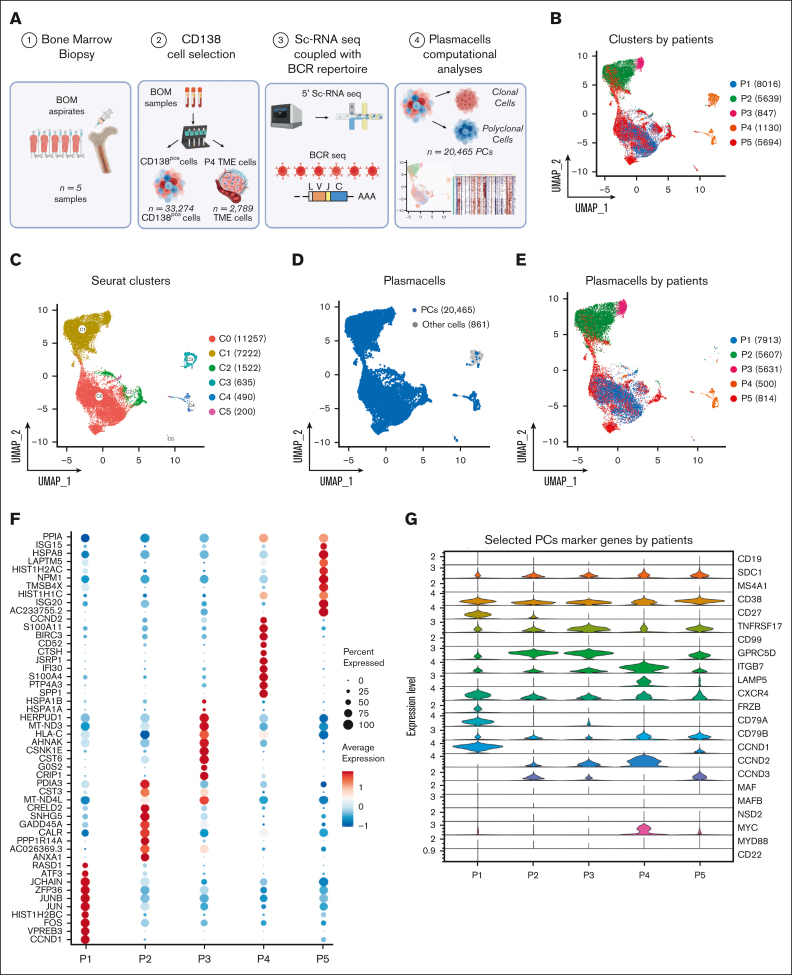
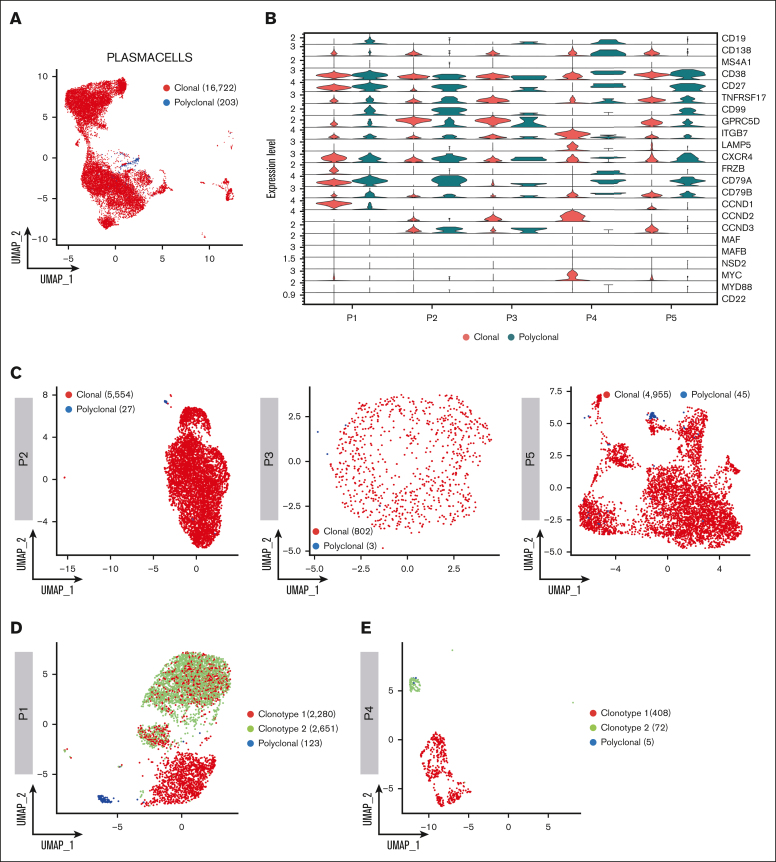
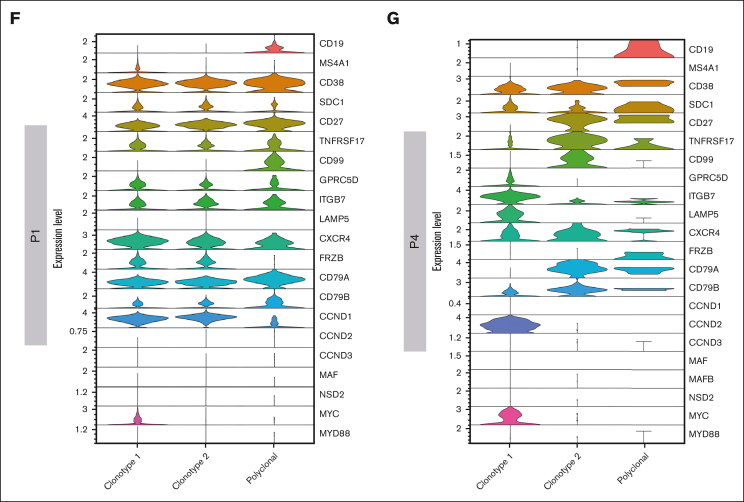
Inference of genomic lesions from single-cell RNA-seq in myeloma improves functional intraclonal and interclonal analysis
- PMID: 38830132
- PMCID: PMC11331727
- DOI: 10.1182/bloodadvances.2023012409
Inference of genomic lesions from single-cell RNA-seq in myeloma improves functional intraclonal and interclonal analysis
Abstract
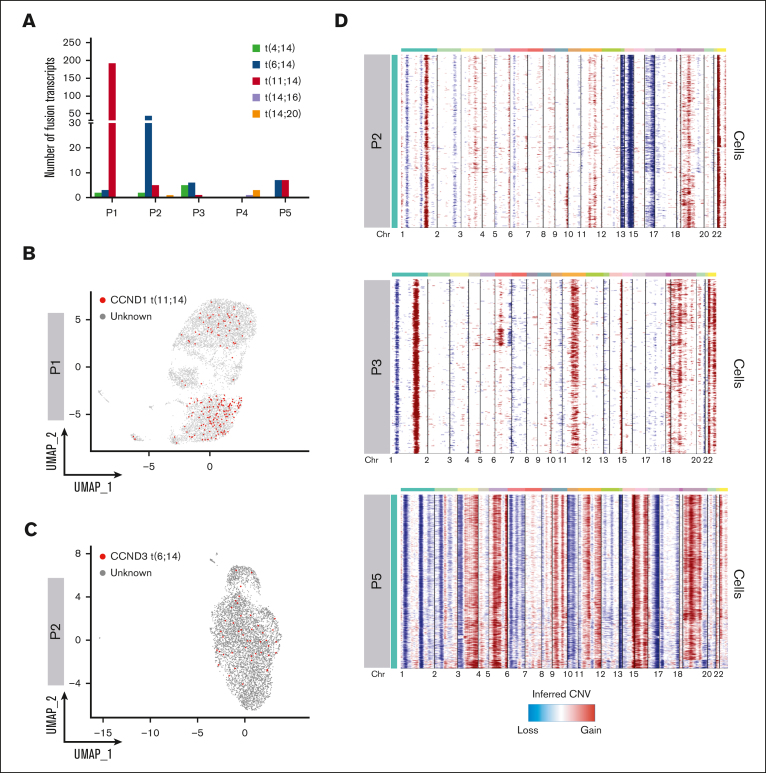
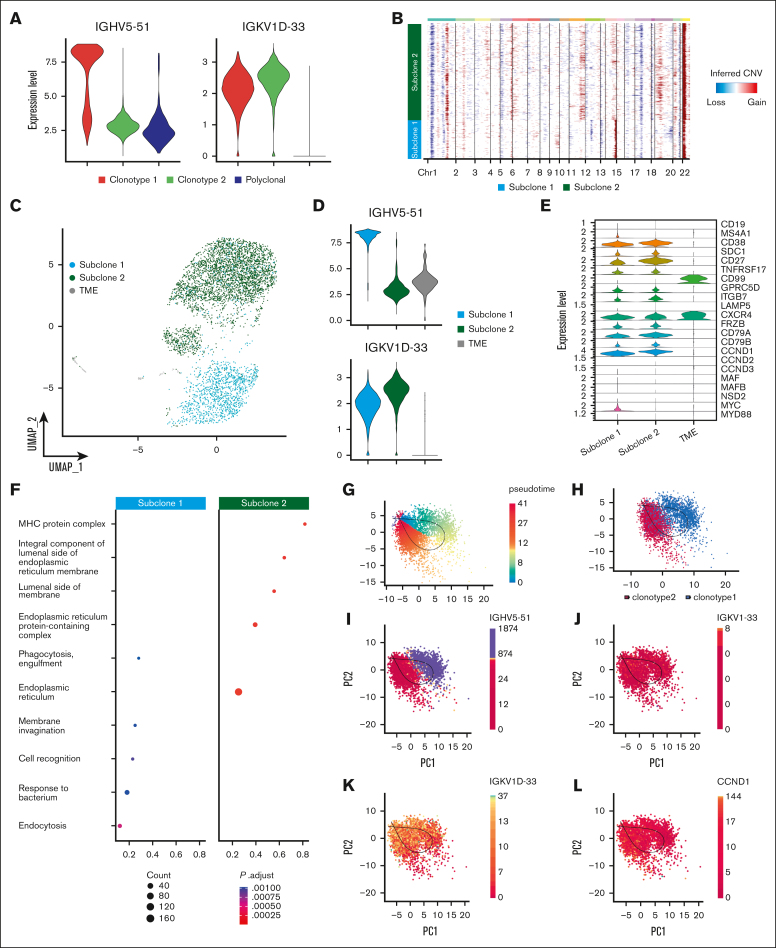
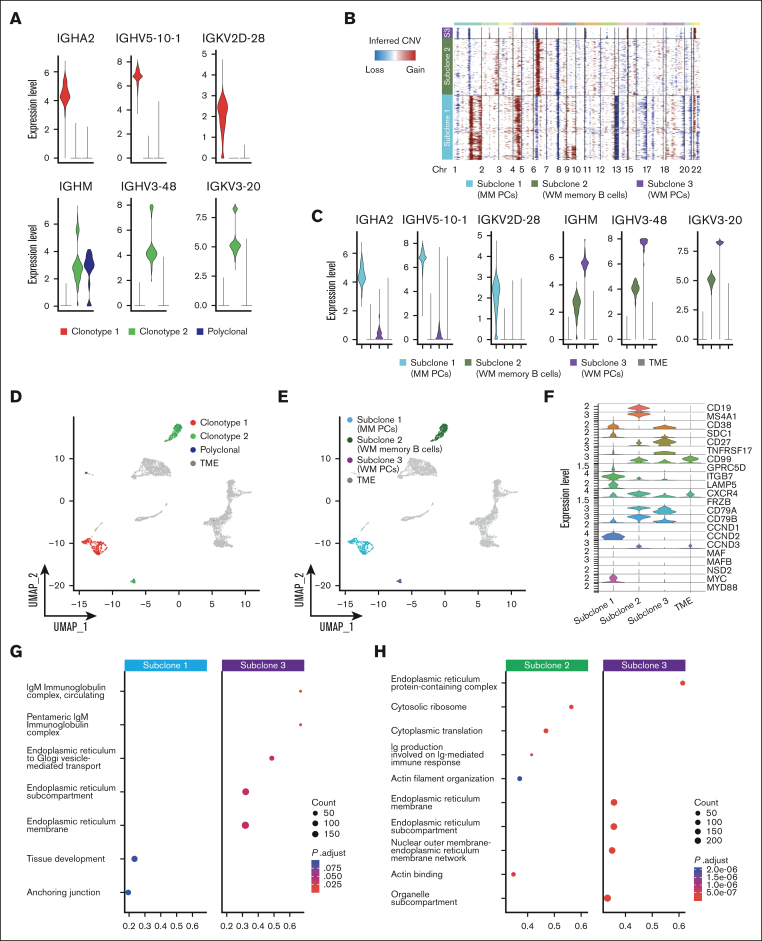
Smoldering multiple myeloma (SMM) is an asymptomatic plasma cell (PC) neoplasm that may evolve with variable frequency into multiple myeloma (MM). SMM is initiated by chromosomal translocations involving the immunoglobulin heavy-chain locus or by hyperdiploidy and evolves through acquisition of additional genetic lesions. In this scenario, we aimed at establishing a reliable analysis pipeline to infer genomic lesions from transcriptomic analysis, by combining single-cell RNA sequencing (scRNA-seq) with B-cell receptor sequencing and copy number abnormality (CNA) analysis to identify clonal PCs at the genetic level along their specific transcriptional landscape. We profiled 20 465 bone marrow PCs derived from 5 patients with SMM/MM and unbiasedly identified clonal and polyclonal PCs. Hyperdiploidy, t(11;14), and t(6;14) were identified at the scRNA level by analysis of chimeric reads. Subclone functional analysis was improved by combining transcriptome with CNA analysis. As examples, we illustrate the different functional properties of a light-chain escape subclone in SMM and of different B-cell and PC subclones in a patient affected by Wäldenstrom macroglobulinemia and SMM. Overall, our data provide a proof of principle for inference of clinically relevant genotypic data from scRNA-seq, which in turn will refine functional annotation of the clonal architecture of PC dyscrasias.
© 2024 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: N.B. received honoraria from Amgen, GlaxoSmithKline (GSK), Janssen, Jazz, Pfizer, and Takeda. M.C.D.V. served on advisory board for Takeda; and speakers bureau for Janssen and GSK. F.P. received honoraria during the last 2 years for lectures from Novartis, Bristol Myers Squibb, AbbVie, GSK, Janssen, and AOP Orphan; and advisory boards fees from Novartis, Bristol Myers Squibb/Celgene, GSK, AbbVie, AOP Orphan, Janssen, Karyopharm, Kyowa Kirin and MEI, Sumitomo, and Kartos. The remaining authors declare no competing financial interests.
Figures