

Longitudinal multicompartment characterization of host-microbiota interactions in patients with acute respiratory failure
- PMID: 38830853
- PMCID: PMC11148165
- DOI: 10.1038/s41467-024-48819-8
Longitudinal multicompartment characterization of host-microbiota interactions in patients with acute respiratory failure
Abstract
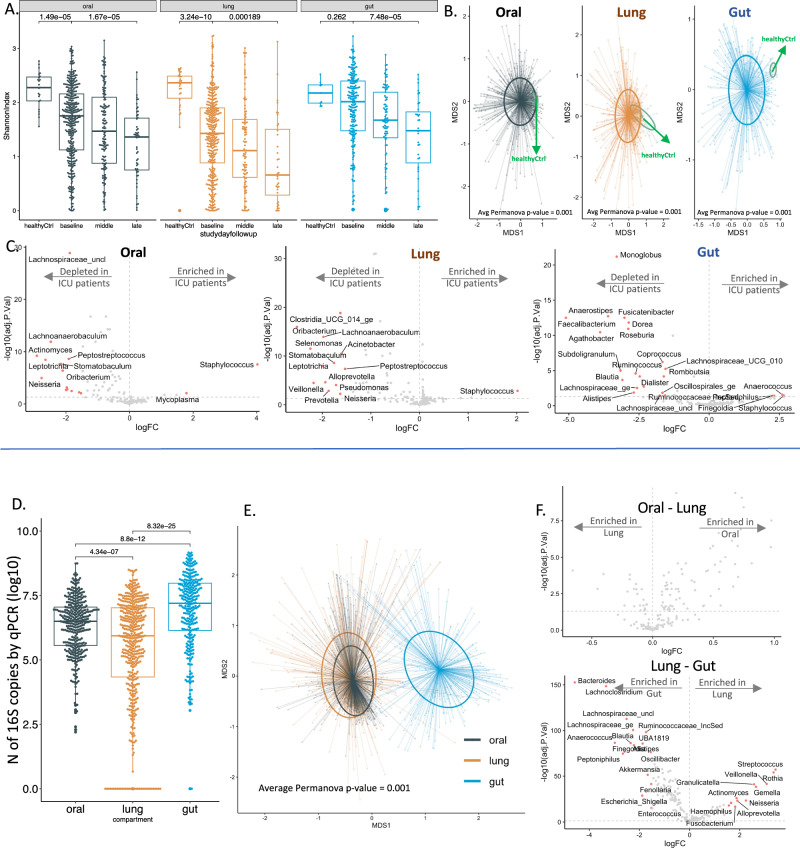
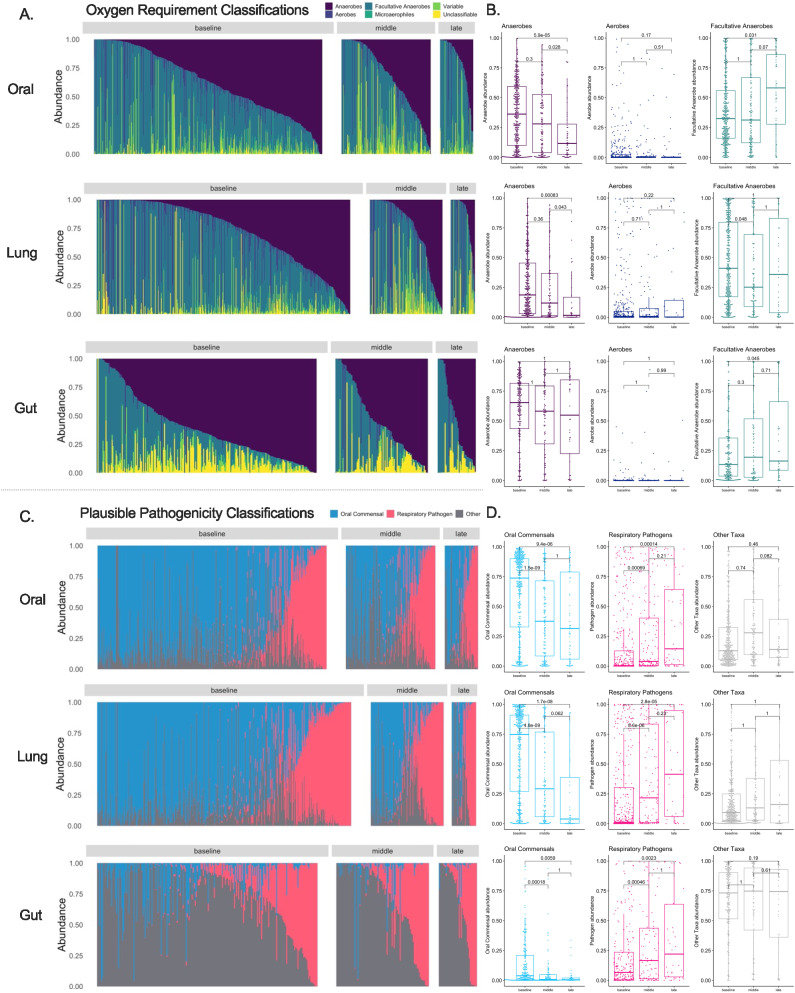
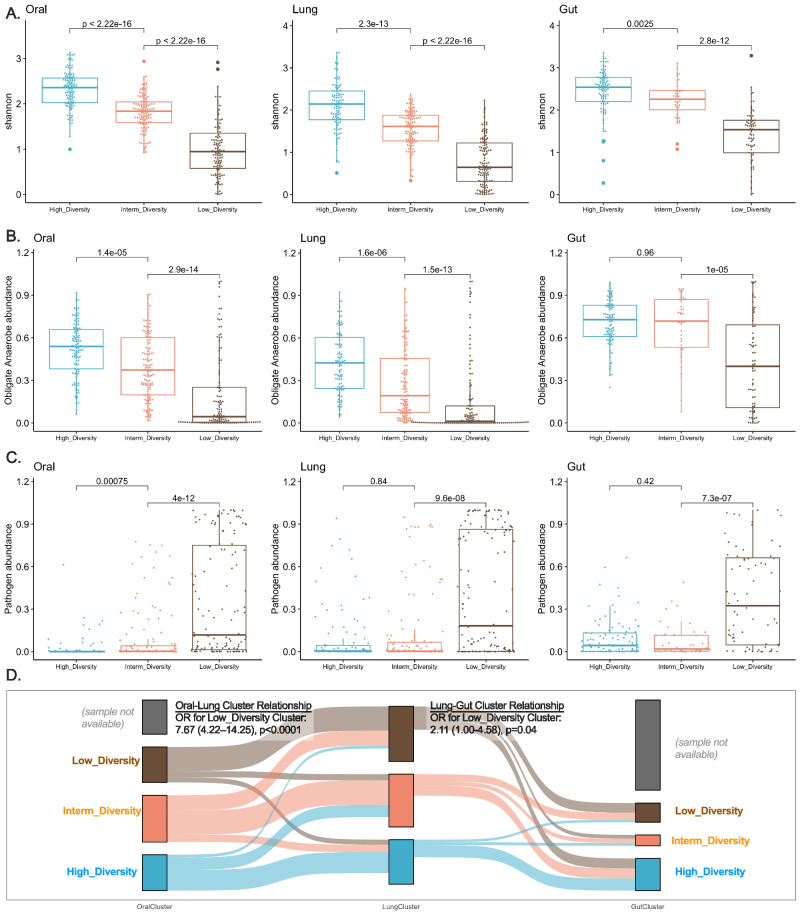
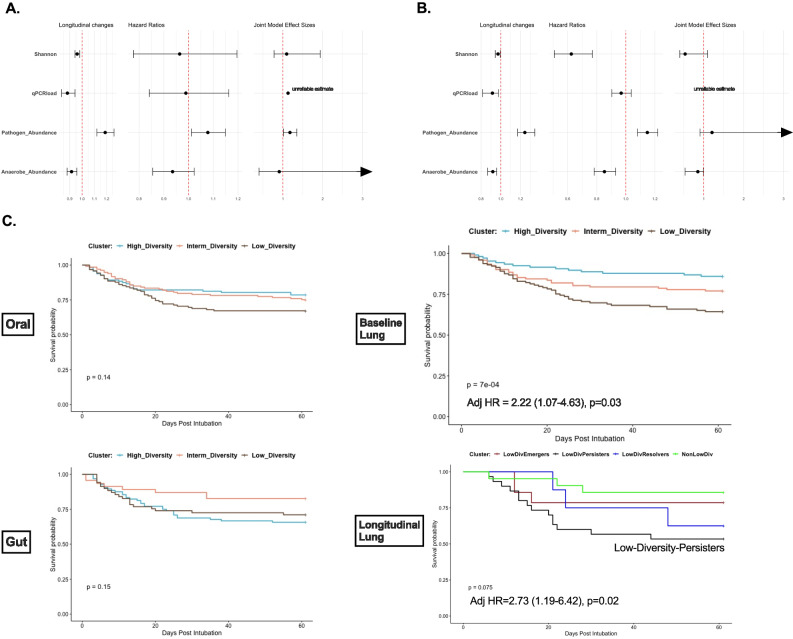
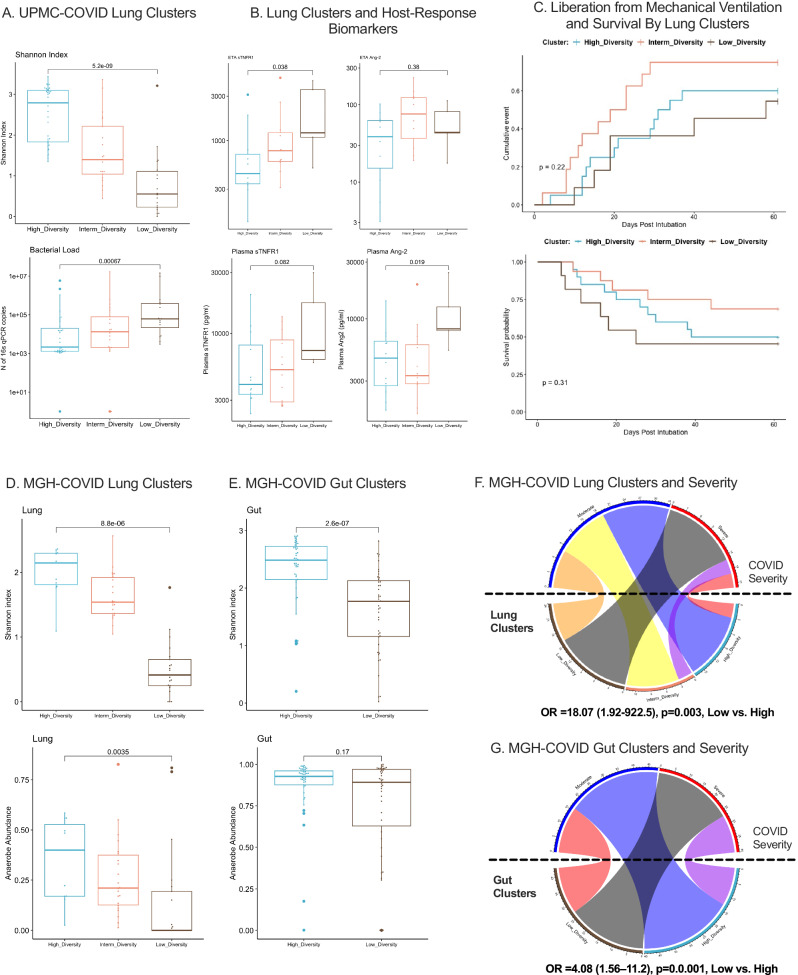
Critical illness can significantly alter the composition and function of the human microbiome, but few studies have examined these changes over time. Here, we conduct a comprehensive analysis of the oral, lung, and gut microbiota in 479 mechanically ventilated patients (223 females, 256 males) with acute respiratory failure. We use advanced DNA sequencing technologies, including Illumina amplicon sequencing (utilizing 16S and ITS rRNA genes for bacteria and fungi, respectively, in all sample types) and Nanopore metagenomics for lung microbiota. Our results reveal a progressive dysbiosis in all three body compartments, characterized by a reduction in microbial diversity, a decrease in beneficial anaerobes, and an increase in pathogens. We find that clinical factors, such as chronic obstructive pulmonary disease, immunosuppression, and antibiotic exposure, are associated with specific patterns of dysbiosis. Interestingly, unsupervised clustering of lung microbiota diversity and composition by 16S independently predicted survival and performed better than traditional clinical and host-response predictors. These observations are validated in two separate cohorts of COVID-19 patients, highlighting the potential of lung microbiota as valuable prognostic biomarkers in critical care. Understanding these microbiome changes during critical illness points to new opportunities for microbiota-targeted precision medicine interventions.
© 2024. The Author(s).
Conflict of interest statement
Dr. Kitsios has received research funding from Karius, Inc, Genentech, Inc, and Pfizer, Inc, all unrelated to this project. Dr. Morris has received research funding from Pfizer, Inc., unrelated to this project. Dr. McVerry has received consulting fees from Boehringer Ingelheim, BioAegis, and Synairgen Research, Ltd. unrelated to this work. All other authors disclosed no conflict of interest.
Figures
Update of
-
Prognostic Insights from Longitudinal Multicompartment Study of Host-Microbiota Interactions in Critically Ill Patients.Res Sq [Preprint]. 2023 Sep 26:rs.3.rs-3338762. doi: 10.21203/rs.3.rs-3338762/v1. Res Sq. 2023. Update in: Nat Commun. 2024 Jun 3;15(1):4708. doi: 10.1038/s41467-024-48819-8. PMID: 37841841 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
