

Environmental activity-based protein profiling for function-driven enzyme discovery from natural communities
- PMID: 38831353
- PMCID: PMC11145796
- DOI: 10.1186/s40793-024-00577-2
Environmental activity-based protein profiling for function-driven enzyme discovery from natural communities
Abstract
Background: Microbial communities are important drivers of global biogeochemical cycles, xenobiotic detoxification, as well as organic matter decomposition. Their major metabolic role in ecosystem functioning is ensured by a unique set of enzymes, providing a tremendous yet mostly hidden enzymatic potential. Exploring this enzymatic repertoire is therefore not only relevant for a better understanding of how microorganisms function in their natural environment, and thus for ecological research, but further turns microbial communities, in particular from extreme habitats, into a valuable resource for the discovery of novel enzymes with potential applications in biotechnology. Different strategies for their uncovering such as bioprospecting, which relies mainly on metagenomic approaches in combination with sequence-based bioinformatic analyses, have emerged; yet accurate function prediction of their proteomes and deciphering the in vivo activity of an enzyme remains challenging.
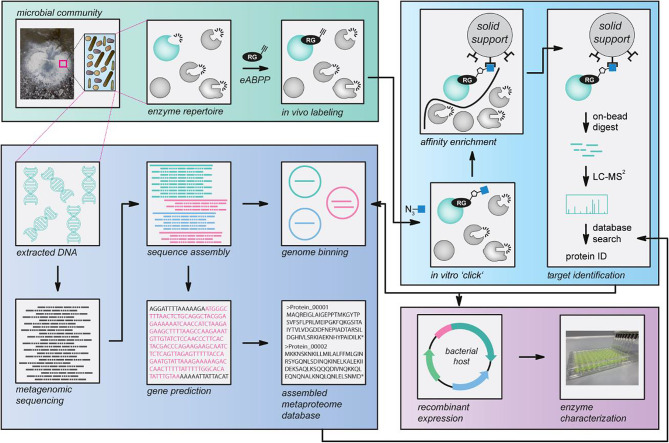
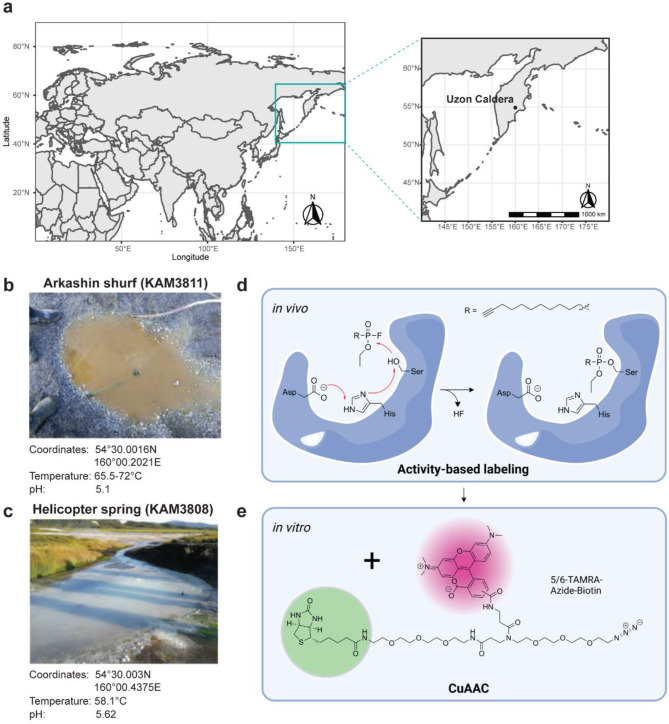
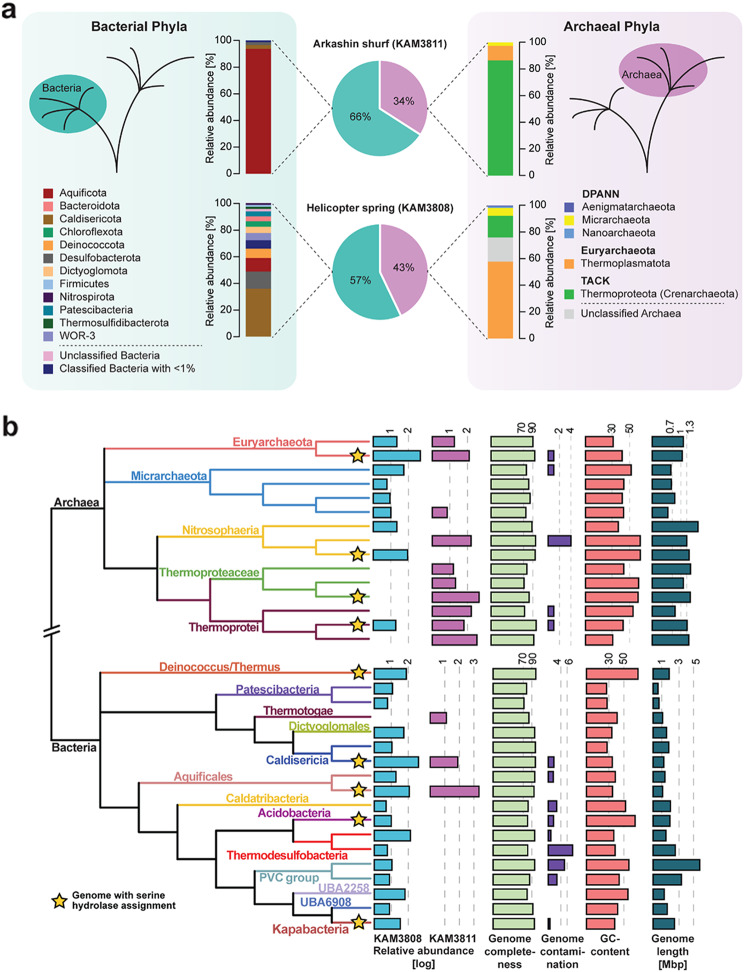
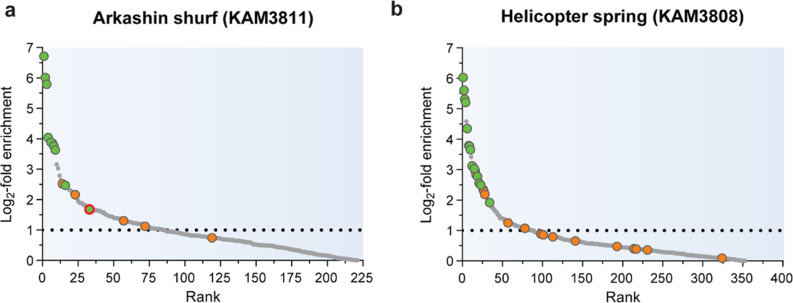
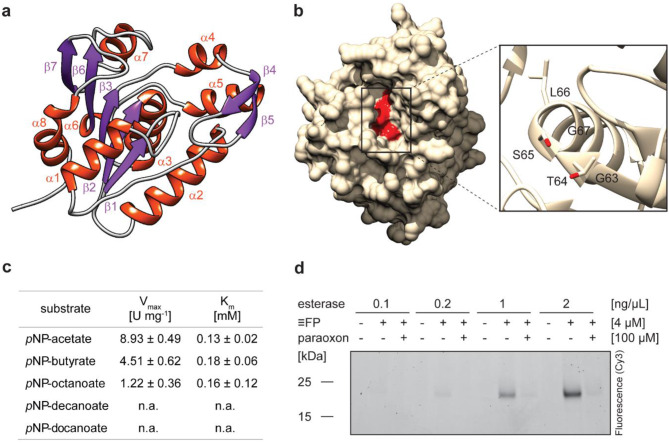
Results: Here, we present environmental activity-based protein profiling (eABPP), a multi-omics approach that extends genome-resolved metagenomics with mass spectrometry-based ABPP. This combination allows direct profiling of environmental community samples in their native habitat and the identification of active enzymes based on their function, even without sequence or structural homologies to annotated enzyme families. eABPP thus bridges the gap between environmental genomics, correct function annotation, and in vivo enzyme activity. As a showcase, we report the successful identification of active thermostable serine hydrolases from eABPP of natural microbial communities from two independent hot springs in Kamchatka, Russia.
Conclusions: By reporting enzyme activities within an ecosystem in their native state, we anticipate that eABPP will not only advance current methodological approaches to sequence homology-guided enzyme discovery from environmental ecosystems for subsequent biocatalyst development but also contributes to the ecological investigation of microbial community interactions by dissecting their underlying molecular mechanisms.
Keywords: Activity-based protein profiling; Chemical proteomics; Click chemistry; Environmental microbial communities; Hot springs; Metagenomics; Metaproteomics; Serine hydrolases; Target identification.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Metaproteomics-guided selection of targeted enzymes for bioprospecting of mixed microbial communities.Biotechnol Biofuels. 2017 May 16;10:128. doi: 10.1186/s13068-017-0815-z. eCollection 2017. Biotechnol Biofuels. 2017. PMID: 28523076 Free PMC article.
-
Unveiling Ecological and Genetic Novelty within Lytic and Lysogenic Viral Communities of Hot Spring Phototrophic Microbial Mats.Microbiol Spectr. 2021 Dec 22;9(3):e0069421. doi: 10.1128/Spectrum.00694-21. Epub 2021 Nov 17. Microbiol Spectr. 2021. PMID: 34787442 Free PMC article.
-
Spatial Metagenomics of Three Geothermal Sites in Pisciarelli Hot Spring Focusing on the Biochemical Resources of the Microbial Consortia.Molecules. 2020 Sep 3;25(17):4023. doi: 10.3390/molecules25174023. Molecules. 2020. PMID: 32899230 Free PMC article.
-
Marine metaproteomics: current status and future directions.J Proteomics. 2014 Jan 31;97:27-35. doi: 10.1016/j.jprot.2013.08.024. Epub 2013 Sep 13. J Proteomics. 2014. PMID: 24041543 Review.
-
Metagenomic analyses: past and future trends.Appl Environ Microbiol. 2011 Feb;77(4):1153-61. doi: 10.1128/AEM.02345-10. Epub 2010 Dec 17. Appl Environ Microbiol. 2011. PMID: 21169428 Free PMC article. Review.
Cited by
-
Harnessing extremophilic carboxylesterases for applications in polyester depolymerisation and plastic waste recycling.Essays Biochem. 2023 Aug 11;67(4):715-729. doi: 10.1042/EBC20220255. Essays Biochem. 2023. PMID: 37334661 Free PMC article.
-
Extremophiles in a changing world.Extremophiles. 2024 Apr 29;28(2):26. doi: 10.1007/s00792-024-01341-7. Extremophiles. 2024. PMID: 38683238 Free PMC article. Review.
-
Characterization of mycobacteria isolated from the Brazilian Atlantic Forest: a public health and bioprospection perspective.Front Microbiol. 2025 Apr 25;16:1558006. doi: 10.3389/fmicb.2025.1558006. eCollection 2025. Front Microbiol. 2025. PMID: 40351310 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous