

Coupled-cluster treatment of complex open-shell systems: the case of single-molecule magnets
- PMID: 38836327
- PMCID: PMC11186456
- DOI: 10.1039/d4cp01129e
Coupled-cluster treatment of complex open-shell systems: the case of single-molecule magnets
Abstract
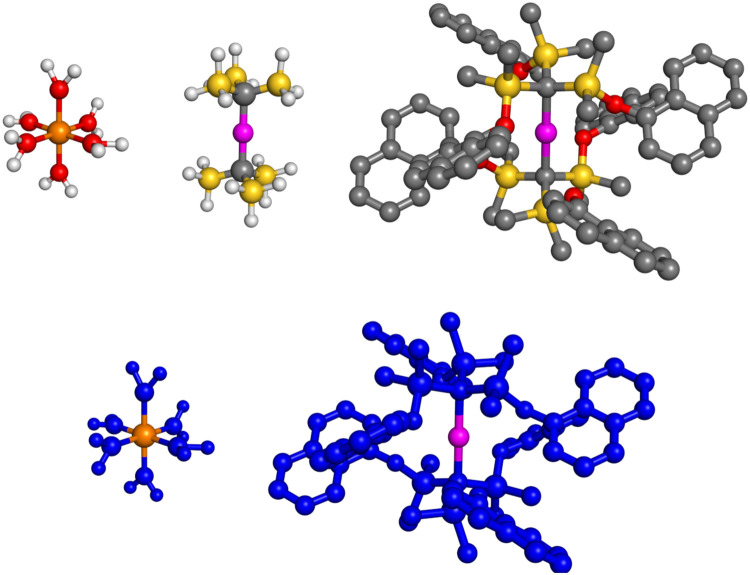
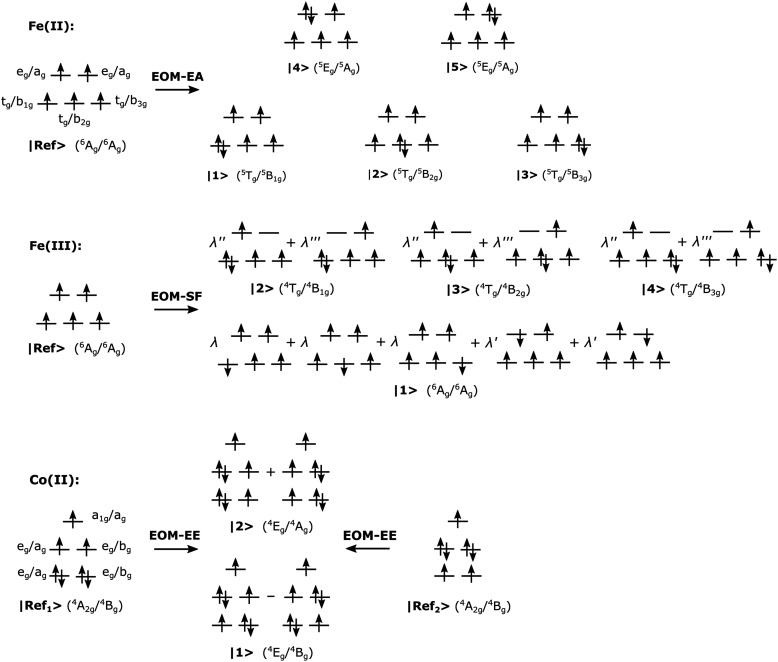
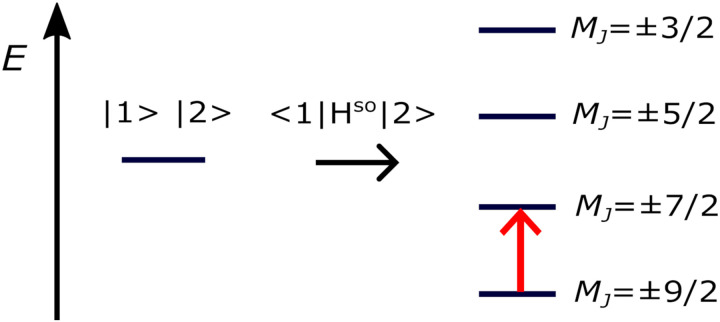
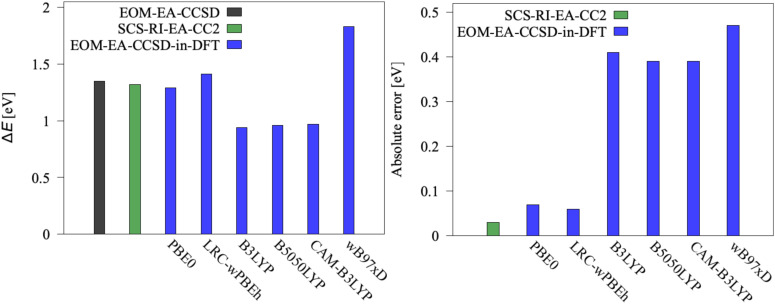
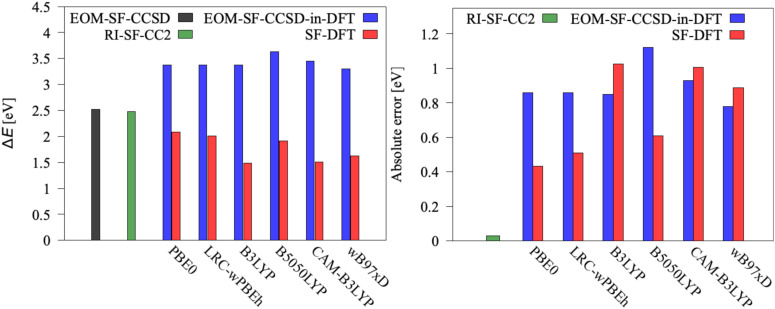
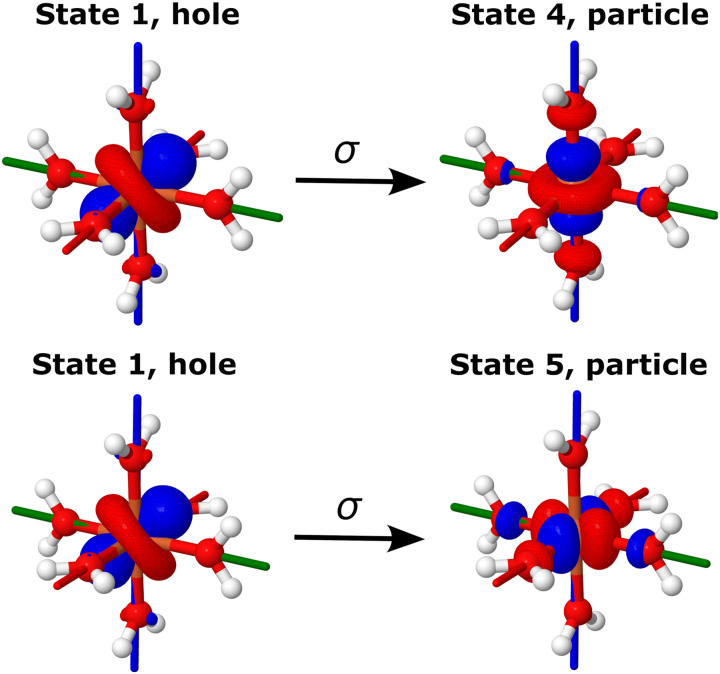
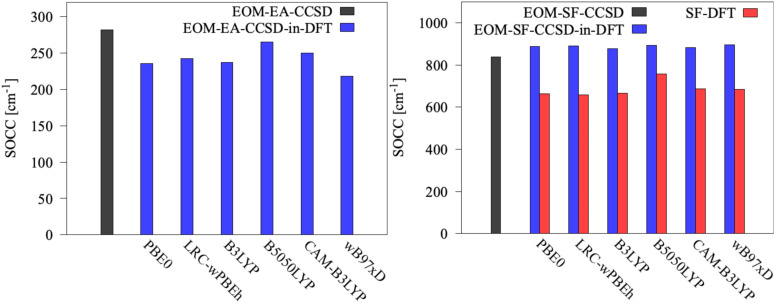
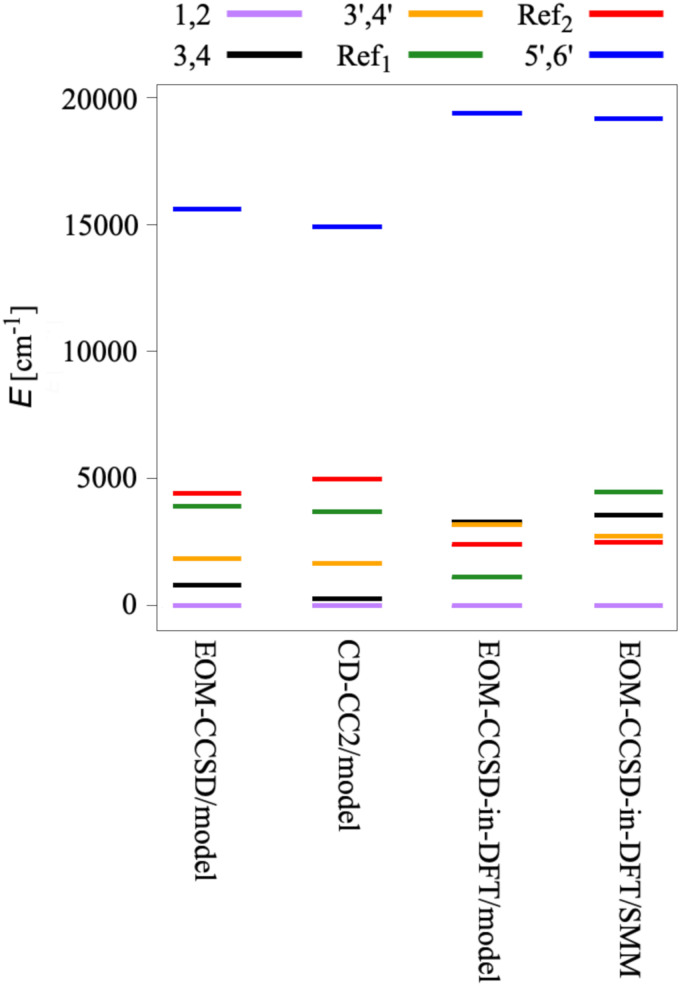
We investigate the reliability of two cost-effective coupled-cluster methods for computing spin-state energetics and spin-related properties of a set of open-shell transition-metal complexes. Specifically, we employ the second-order approximate coupled-cluster singles and doubles (CC2) method and projection-based embedding that combines equation-of-motion coupled-cluster singles and doubles (EOM-CCSD) with density functional theory (DFT). The performance of CC2 and EOM-CCSD-in-DFT is assessed against EOM-CCSD. The chosen test set includes two hexaaqua transition-metal complexes containing Fe(II) and Fe(III), and a large Co(II)-based single-molecule magnet with a non-aufbau ground state. We find that CC2 describes the excited states more accurately, reproducing EOM-CCSD excitation energies within 0.05 eV. However, EOM-CCSD-in-DFT excels in describing transition orbital angular momenta and spin-orbit couplings. Moreover, for the Co(II) molecular magnet, using EOM-CCSD-in-DFT eigenstates and spin-orbit couplings, we compute spin-reversal energy barriers, as well as temperature-dependent and field-dependent magnetizations and magnetic susceptibilities that closely match experimental values within spectroscopic accuracy. These results underscore the efficiency of CC2 in computing state energies of multi-configurational, open-shell systems and highlight the utility of the more cost-efficient EOM-CCSD-in-DFT for computing spin-orbit couplings and magnetic properties of complex and large molecular magnets.
Conflict of interest statement
There are no conflicts to declare.
Figures
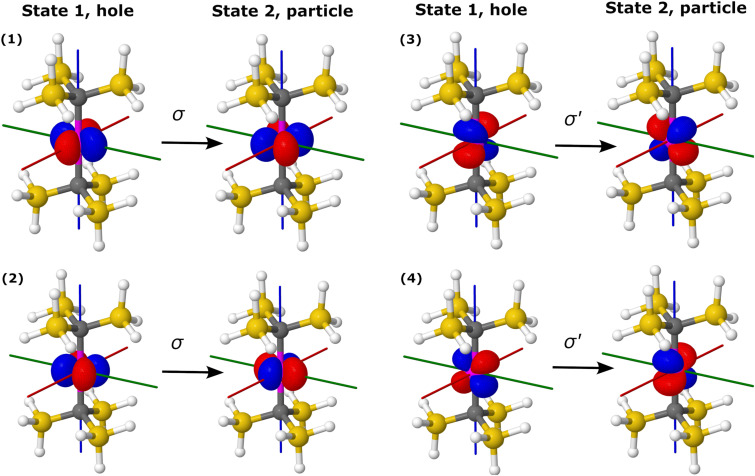
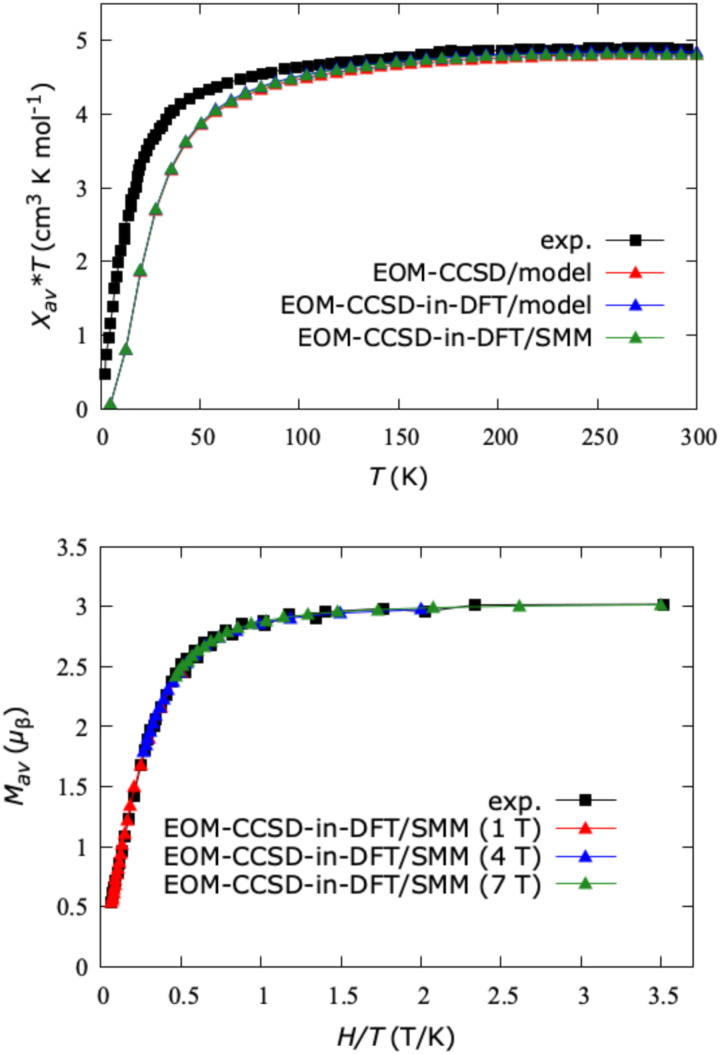
Similar articles
-
Quantum-embedded equation-of-motion coupled-cluster approach to single-atom magnets on surfaces.Phys Chem Chem Phys. 2025 Jul 23;27(29):15474-15485. doi: 10.1039/d5cp01059d. Phys Chem Chem Phys. 2025. PMID: 40624939 Free PMC article.
-
Assessment of low-scaling approximations to the equation of motion coupled-cluster singles and doubles equations.J Chem Phys. 2014 Oct 28;141(16):164116. doi: 10.1063/1.4898709. J Chem Phys. 2014. PMID: 25362281
-
Combining the spin-separated exact two-component relativistic Hamiltonian with the equation-of-motion coupled-cluster method for the treatment of spin-orbit splittings of light and heavy elements.Phys Chem Chem Phys. 2017 Feb 1;19(5):3713-3721. doi: 10.1039/c6cp07588f. Phys Chem Chem Phys. 2017. PMID: 28097277
-
Equation-of-Motion Coupled-Cluster Theory for Excitation Energies of Closed-Shell Systems with Spin-Orbit Coupling.J Chem Theory Comput. 2014 Dec 9;10(12):5567-76. doi: 10.1021/ct500854m. J Chem Theory Comput. 2014. PMID: 26583239
-
Understanding the Spin of Metal Complexes from a Single-Molecule Perspective.Small Methods. 2025 May;9(5):e2401302. doi: 10.1002/smtd.202401302. Epub 2024 Nov 10. Small Methods. 2025. PMID: 39523749 Review.
Cited by
-
Quantum-embedded equation-of-motion coupled-cluster approach to single-atom magnets on surfaces.Phys Chem Chem Phys. 2025 Jul 23;27(29):15474-15485. doi: 10.1039/d5cp01059d. Phys Chem Chem Phys. 2025. PMID: 40624939 Free PMC article.
References
LinkOut - more resources
Full Text Sources