

IRE1α Mediates the Hypertrophic Growth of Cardiomyocytes Through Facilitating the Formation of Initiation Complex to Promote the Translation of TOP-Motif Transcripts
- PMID: 38836349
- PMCID: PMC11427172
- DOI: 10.1161/CIRCULATIONAHA.123.067606
IRE1α Mediates the Hypertrophic Growth of Cardiomyocytes Through Facilitating the Formation of Initiation Complex to Promote the Translation of TOP-Motif Transcripts
Abstract
Background: Cardiomyocyte growth is coupled with active protein synthesis, which is one of the basic biological processes in living cells. However, it is unclear whether the unfolded protein response transducers and effectors directly take part in the control of protein synthesis. The connection between critical functions of the unfolded protein response in cellular physiology and requirements of multiple processes for cell growth prompted us to investigate the role of the unfolded protein response in cell growth and underlying molecular mechanisms.
Methods: Cardiomyocyte-specific inositol-requiring enzyme 1α (IRE1α) knockout and overexpression mouse models were generated to explore its function in vivo. Neonatal rat ventricular myocytes were isolated and cultured to evaluate the role of IRE1α in cardiomyocyte growth in vitro. Mass spectrometry was conducted to identify novel interacting proteins of IRE1α. Ribosome sequencing and polysome profiling were performed to determine the molecular basis for the function of IRE1α in translational control.
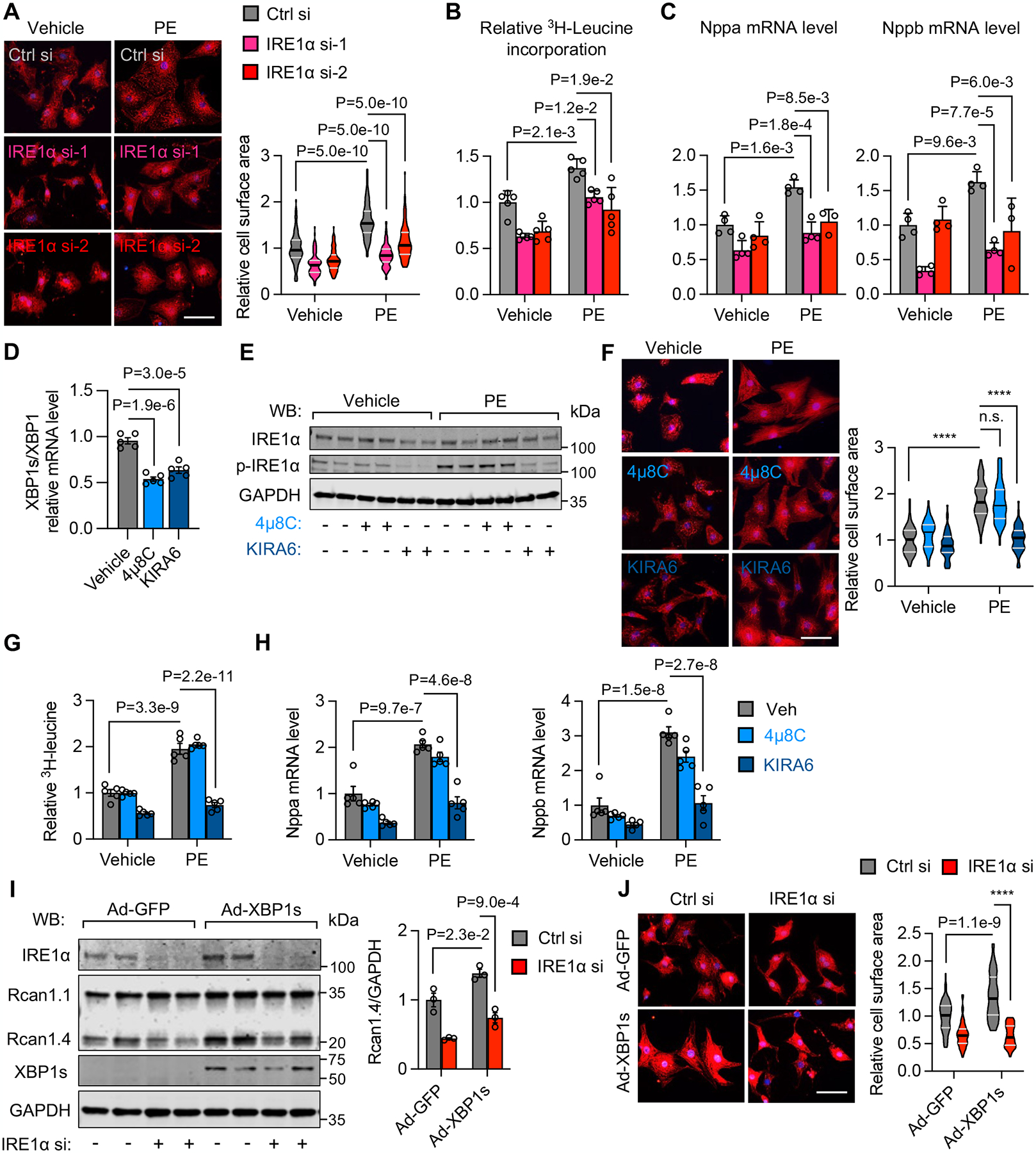
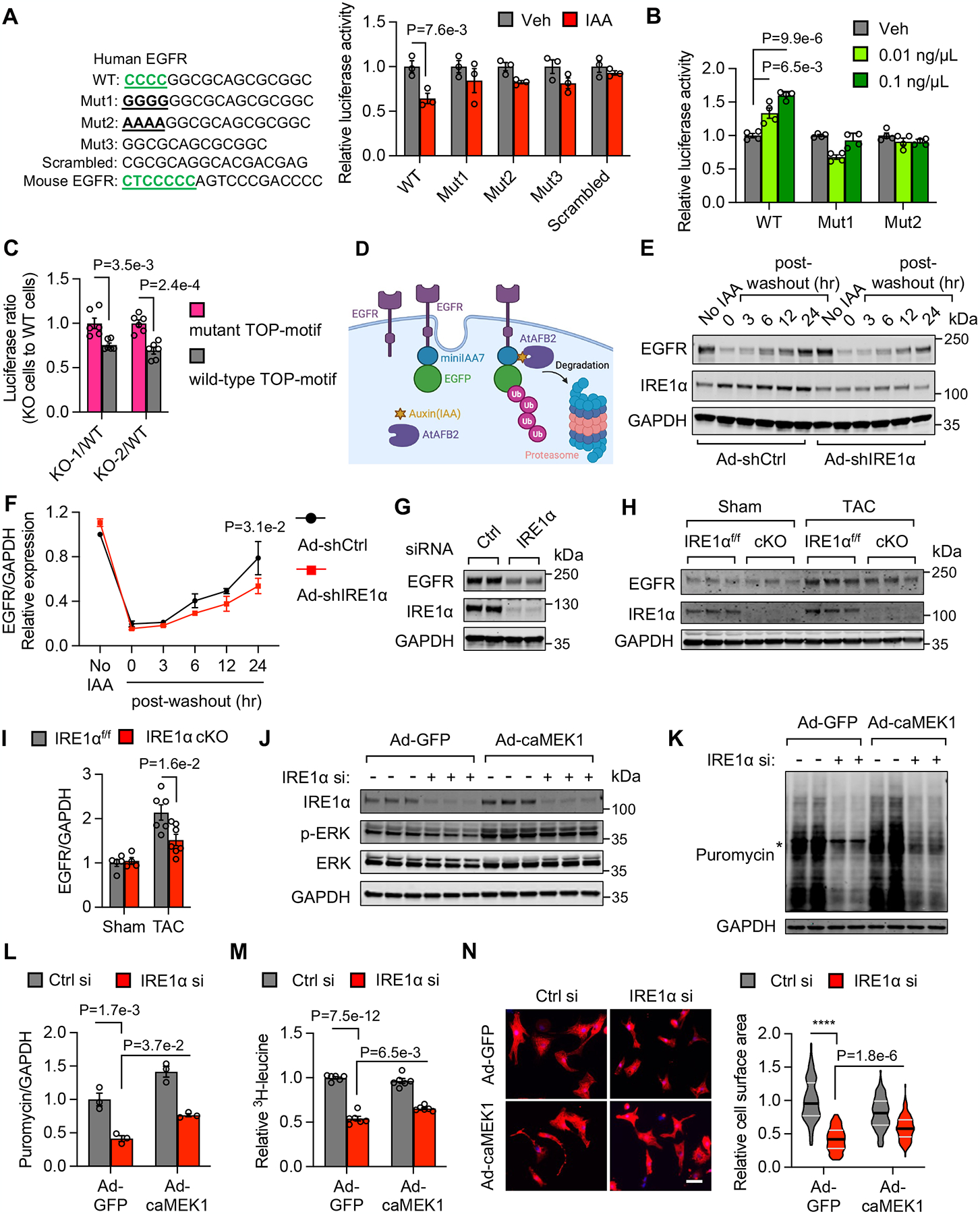
Results: We show that IRE1α is required for cell growth in neonatal rat ventricular myocytes under prohypertrophy treatment and in HEK293 cells in response to serum stimulation. At the molecular level, IRE1α directly interacts with eIF4G and eIF3, 2 critical components of the translation initiation complex. We demonstrate that IRE1α facilitates the formation of the translation initiation complex around the endoplasmic reticulum and preferentially initiates the translation of transcripts with 5' terminal oligopyrimidine motifs. We then reveal that IRE1α plays an important role in determining the selectivity and translation of these transcripts. We next show that IRE1α stimulates the translation of epidermal growth factor receptor through an unannotated terminal oligopyrimidine motif in its 5' untranslated region. We further demonstrate a physiological role of IRE1α-governed protein translation by showing that IRE1α is essential for cardiomyocyte growth and cardiac functional maintenance under hemodynamic stress in vivo.
Conclusions: These studies suggest a noncanonical, essential role of IRE1α in orchestrating protein synthesis, which may have important implications in cardiac hypertrophy in response to pressure overload and general cell growth under other physiological and pathological conditions.
Keywords: EGFR protein; eukaryonic initiation factor; heart failure; unfolded protein response.
Conflict of interest statement
None.
Figures





References
-
- Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK, Buxton AE, et al. Heart disease and stroke statistics-2023 update: A report from the American Heart Association. Circulation. 2023;147:e93–e621. doi: 10.1161/cir.0000000000001123 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
