

Late-onset Krabbe disease presenting as spastic paraplegia - implications of GCase and CTSB/D
- PMID: 38837642
- PMCID: PMC11251474
- DOI: 10.1002/acn3.52078
Late-onset Krabbe disease presenting as spastic paraplegia - implications of GCase and CTSB/D
Abstract
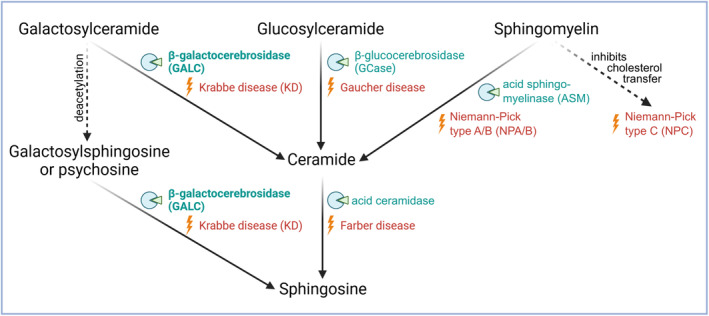
Objective: Krabbe disease (KD) is a multisystem neurodegenerative disorder with severe disability and premature death, mostly with an infancy/childhood onset. In rare cases of late-onset phenotypes, symptoms are often milder and difficult to diagnose. We here present a translational approach combining diagnostic and biochemical analyses of a male patient with a progressive gait disorder starting at the age of 44 years, with a final diagnosis of late-onset KD (LOKD).
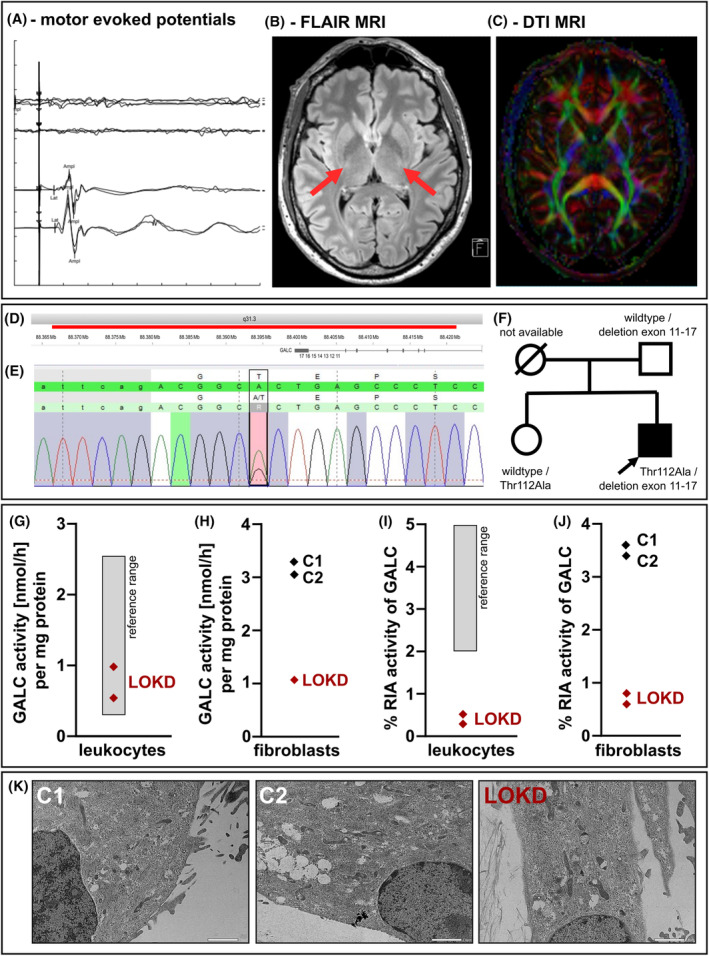
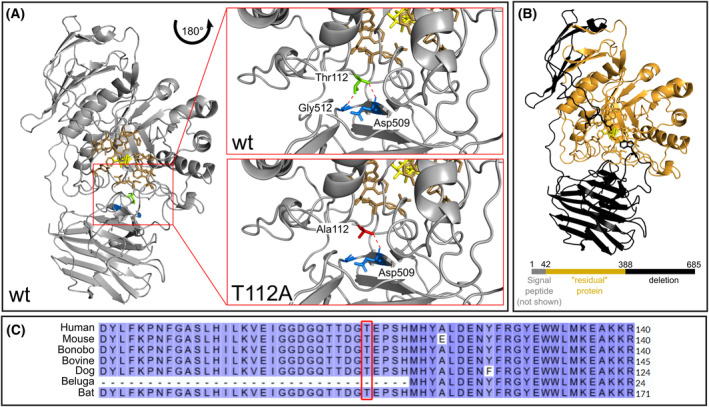
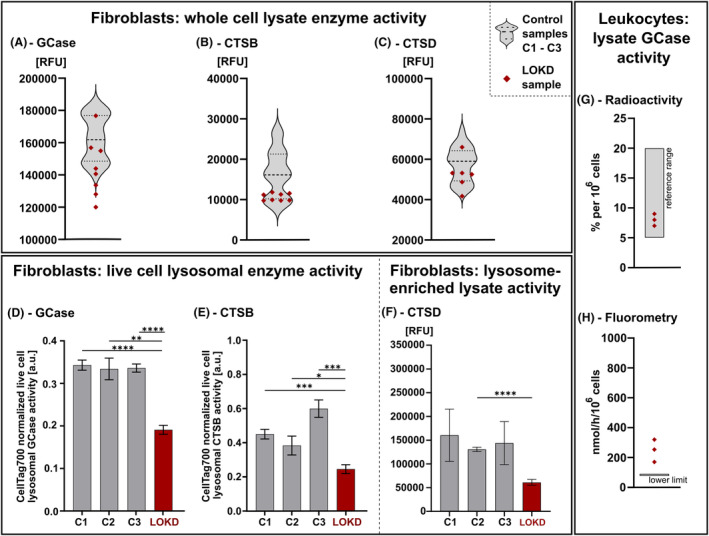
Methods: Additionally to cerebral MRI, protein structural analyses of the β-galactocerebrosidase protein (GALC) were performed. Moreover, expression, lysosomal localization, and activities of β-glucocerebrosidase (GCase), cathepsin B (CTSB), and cathepsin D (CTSD) were analyzed in leukocytes, fibroblasts, and lysosomes of fibroblasts.
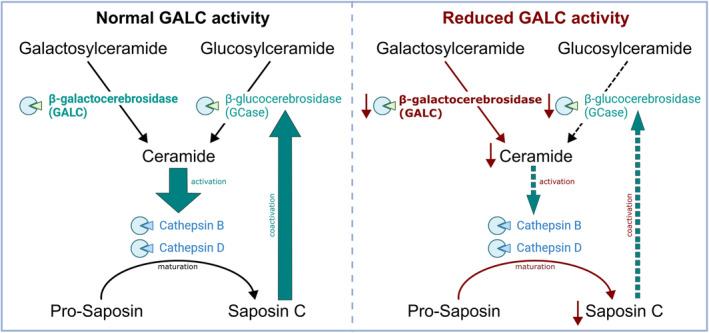
Results: Exome sequencing revealed biallelic likely pathogenic variants: GALC exons 11-17: 33 kb deletion; exon 4: missense variant (c.334A>G, p.Thr112Ala). We detected a reduced GALC activity in leukocytes and fibroblasts. While histological KD phenotypes were absent in fibroblasts, they showed a significantly decreased activities of GCase, CTSB, and CTSD in lysosomal fractions, while expression levels were unaffected.
Interpretation: The presented LOKD case underlines the age-dependent appearance of a mildly pathogenic GALC variant and its interplay with other lysosomal proteins. As GALC malfunction results in reduced ceramide levels, we assume this to be causative for the here described decrease in CTSB and CTSD activity, potentially leading to diminished GCase activity. Hence, we emphasize the importance of a functional interplay between the lysosomal enzymes GALC, CTSB, CTSD, and GCase, as well as between their substrates, and propose their conjoined contribution in KD pathology.
Keywords: Krabbe disease; cathepsin B; cathepsin D; enzymatic activity; late‐onset; β‐glucocerebrosidase.
© 2024 The Authors. Annals of Clinical and Translational Neurology published by Wiley Periodicals LLC on behalf of American Neurological Association.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures

References
-
- Heim P, Claussen M, Hoffmann B, et al. Leukodystrophy incidence in Germany. Am J Med Genet. 1997;71:475‐478. - PubMed
-
- Harzer K, Knoblich R, Rolfs A, Bauer P, Eggers J. Residual galactosylsphingosine (psychosine) β‐galactosidase activities and associated GALC mutations in late and very late onset Krabbe disease. Clin Chim Acta. 2002;317:77‐84. - PubMed
-
- Duffner PK, Barczykowski A, Kay DM, et al. Later onset phenotypes of Krabbe disease: results of the world‐wide registry. Pediatr Neurol. 2012;46:298‐306. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- N8/Interdisciplinary Center for Clinical Research (IZKF) of the University Hospital of the University of Erlangen-Nuremberg
- 01GM1905B/Bundesministerium für Bildung und Forschung (BMBF)
- 01GM2205B/Bundesministerium für Bildung und Forschung (BMBF)
- 01EO2105/Bundesministerium für Bildung und Forschung (BMBF)
- 270949263/GRK2162/Deutsche Forschungsgemeinschaft
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
