

Low-dose aspirin for the prevention of atherosclerotic cardiovascular disease
- PMID: 38839268
- PMCID: PMC11242460
- DOI: 10.1093/eurheartj/ehae324
Low-dose aspirin for the prevention of atherosclerotic cardiovascular disease
Abstract
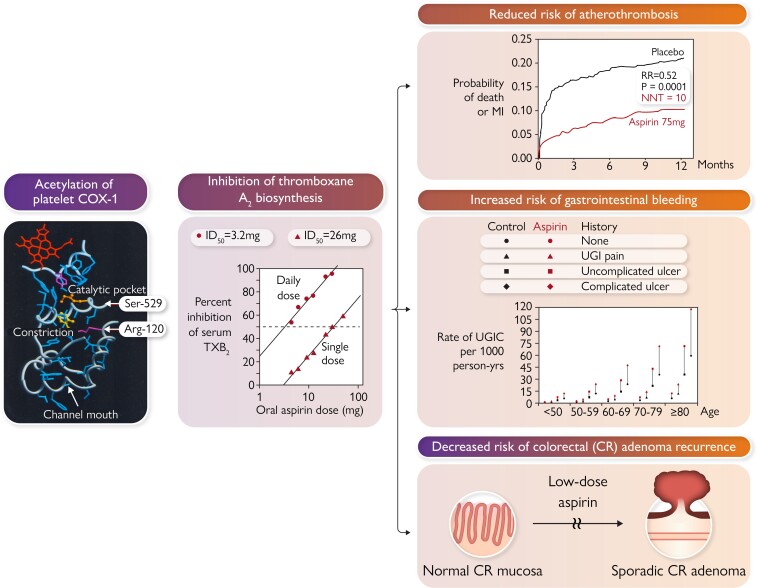
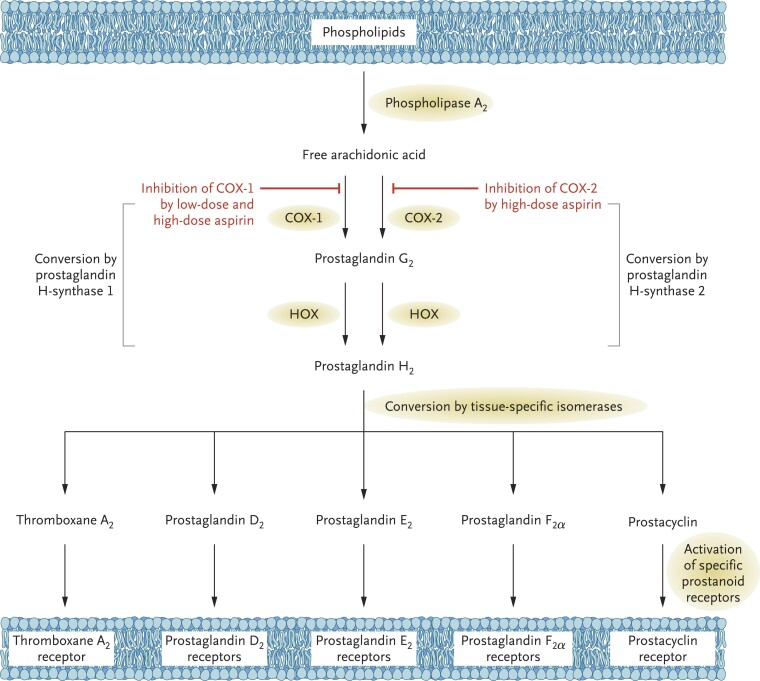
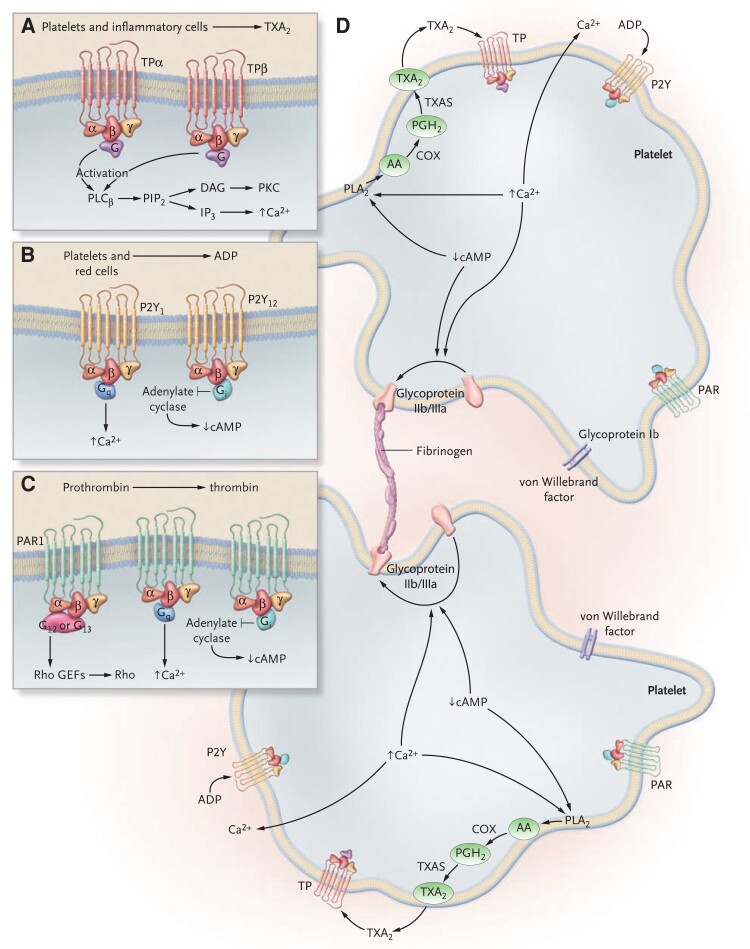
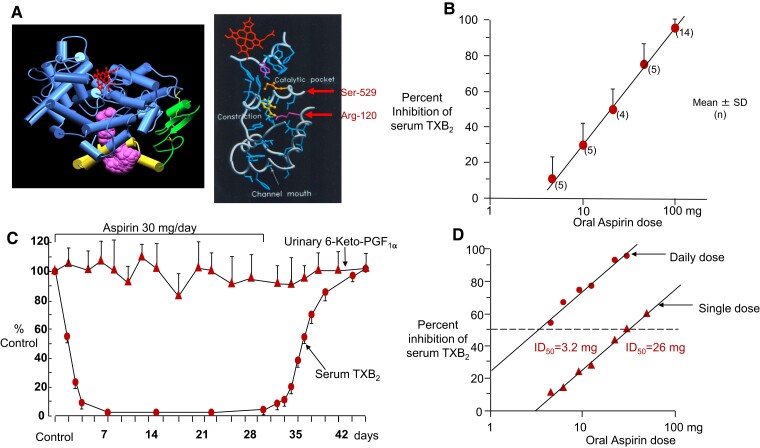
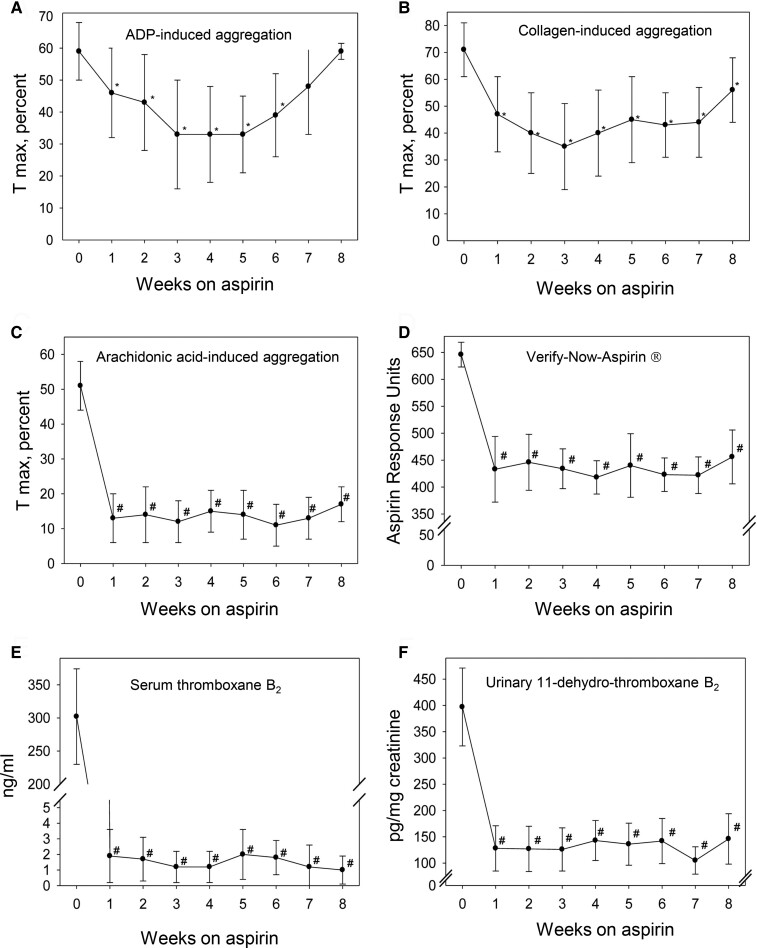
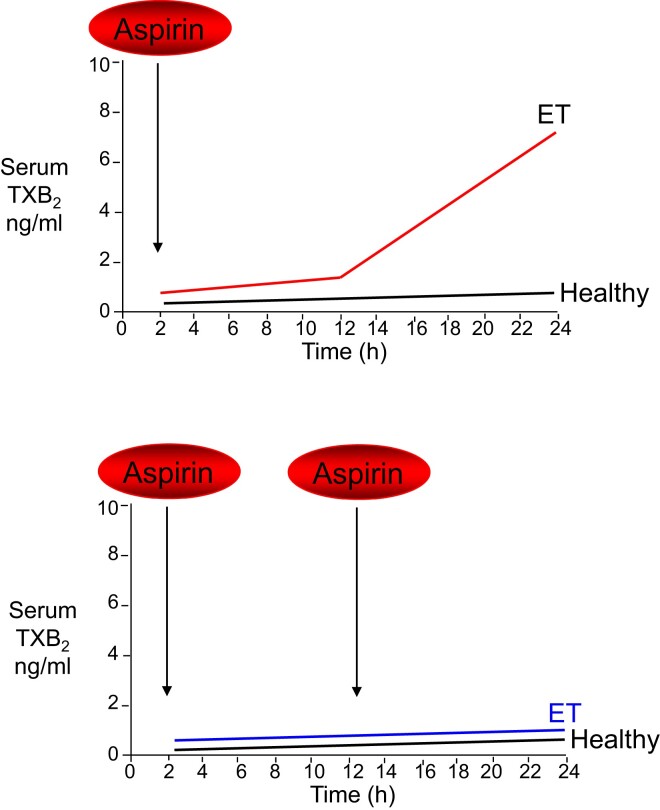
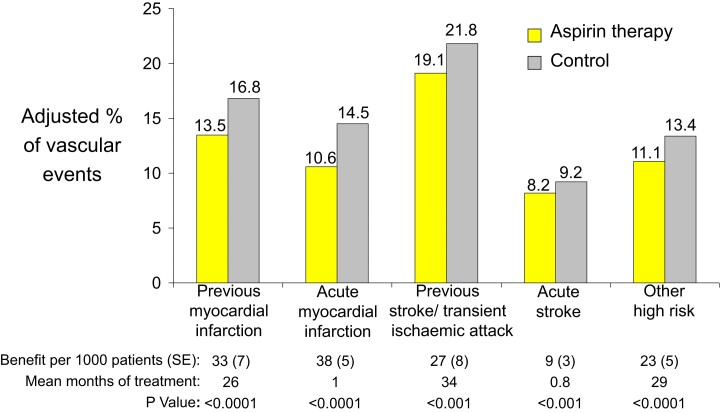
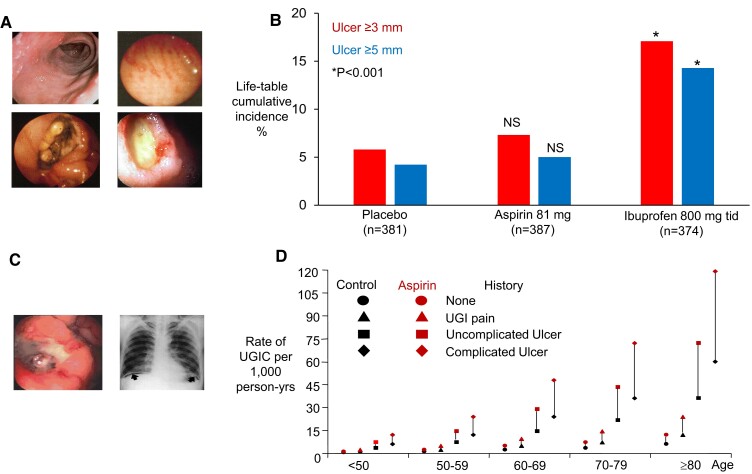
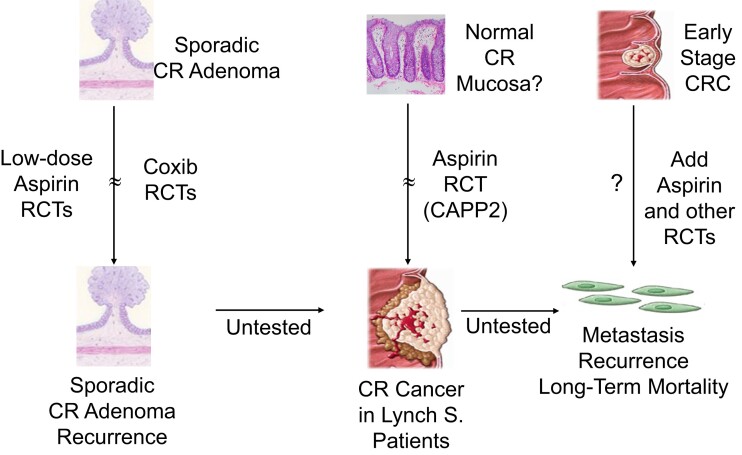
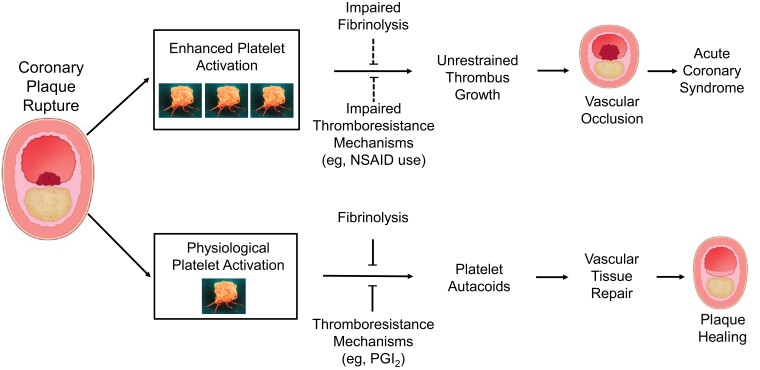
During the past 30 years, several developments have occurred in the antiplatelet field, including the role of aspirin in primary prevention of atherosclerotic cardiovascular disease. There have been several attempts to develop antiplatelet drugs more effective and safer than aspirin and a shift in emphasis from efficacy to safety, advocating aspirin-free antiplatelet regimens after percutaneous coronary intervention. Evidence supporting a chemopreventive effect of low-dose aspirin against colorectal (and other digestive tract) cancer has also strengthened. The aim of this article is to revisit the role of aspirin in the prevention of atherothrombosis across the cardiovascular risk continuum, in view of developments in the antiplatelet field. The review will offer a clinical perspective on aspirin's mechanism of action, pharmacokinetics, and pharmacodynamics. This will be followed by a detailed discussion of its clinical efficacy and safety.
Keywords: Antiplatelet therapy; Aspirin; Atherosclerotic cardiovascular disease; Atherothrombosis; Bleeding; Clopidogrel; Colorectal cancer; P2Y12 inhibitors; Ticagrelor.
© The Author(s) 2024. Published by Oxford University Press on behalf of the European Society of Cardiology.
Figures
Similar articles
-
Role of and Recent Evidence for Antiplatelet Therapy in Prevention of Cardiovascular Disease in Diabetes.Curr Cardiol Rep. 2019 Jun 28;21(8):78. doi: 10.1007/s11886-019-1168-y. Curr Cardiol Rep. 2019. PMID: 31254105 Review.
-
Effects of P2Y12 receptor inhibition with or without aspirin on hemostatic system activation: a randomized trial in healthy subjects.J Thromb Haemost. 2016 Feb;14(2):273-81. doi: 10.1111/jth.13216. Epub 2016 Jan 30. J Thromb Haemost. 2016. PMID: 26663880 Clinical Trial.
-
Combined antiplatelet therapy: still a sweeping combination in cardiology.Cardiovasc Hematol Agents Med Chem. 2013 Jun;11(2):136-67. doi: 10.2174/1871525711311020009. Cardiovasc Hematol Agents Med Chem. 2013. PMID: 23597106 Review.
-
Antiplatelet drugs--do we need new options? With a reappraisal of direct thromboxane inhibitors.Drugs. 2010 May 7;70(7):887-908. doi: 10.2165/11536000-000000000-00000. Drugs. 2010. PMID: 20426498 Review.
-
Ticagrelor plus aspirin for 1 month, followed by ticagrelor monotherapy for 23 months vs aspirin plus clopidogrel or ticagrelor for 12 months, followed by aspirin monotherapy for 12 months after implantation of a drug-eluting stent: a multicentre, open-label, randomised superiority trial.Lancet. 2018 Sep 15;392(10151):940-949. doi: 10.1016/S0140-6736(18)31858-0. Epub 2018 Aug 27. Lancet. 2018. PMID: 30166073 Clinical Trial.
Cited by
-
Role of aspirin in the primary prevention of cardiovascular disease in patients with hyperlipoproteinaemia(a).Drugs Context. 2024 Dec 31;13:2024-10-2. doi: 10.7573/dic.2024-10-2. eCollection 2024. Drugs Context. 2024. PMID: 39763632 Free PMC article. Review.
-
Investigating the role of aspirin on the mortality risk of sepsis-associated encephalopathy: a retrospective study.Front Neurol. 2025 Mar 26;16:1521043. doi: 10.3389/fneur.2025.1521043. eCollection 2025. Front Neurol. 2025. PMID: 40206292 Free PMC article.
-
A review of natural compounds to regulate platelet aggregation: molecular mechanism and research advance.Front Pharmacol. 2025 Jun 27;16:1537776. doi: 10.3389/fphar.2025.1537776. eCollection 2025. Front Pharmacol. 2025. PMID: 40657646 Free PMC article. Review.
-
Myeloproliferative Neoplasms and Cardiovascular Disease: A Review.Curr Treat Options Oncol. 2024 Oct;25(10):1257-1267. doi: 10.1007/s11864-024-01255-8. Epub 2024 Sep 16. Curr Treat Options Oncol. 2024. PMID: 39278999 Review.
-
Population cessation of aspirin use for the prevention of cardiovascular disease.Am J Prev Cardiol. 2025 Feb 8;21:100941. doi: 10.1016/j.ajpc.2025.100941. eCollection 2025 Mar. Am J Prev Cardiol. 2025. PMID: 40103687 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
