

Defining the KRAS- and ERK-dependent transcriptome in KRAS-mutant cancers
- PMID: 38843331
- PMCID: PMC11301402
- DOI: 10.1126/science.adk0775
Defining the KRAS- and ERK-dependent transcriptome in KRAS-mutant cancers
Erratum in
-
Erratum for the Research Article "Defining the KRAS- and ERK-dependent transcriptome in KRAS-mutant cancers" by J. A. Klomp et al.Science. 2024 Aug 23;385(6711):eads4435. doi: 10.1126/science.ads4435. Epub 2024 Aug 22. Science. 2024. PMID: 39172854 No abstract available.
Abstract
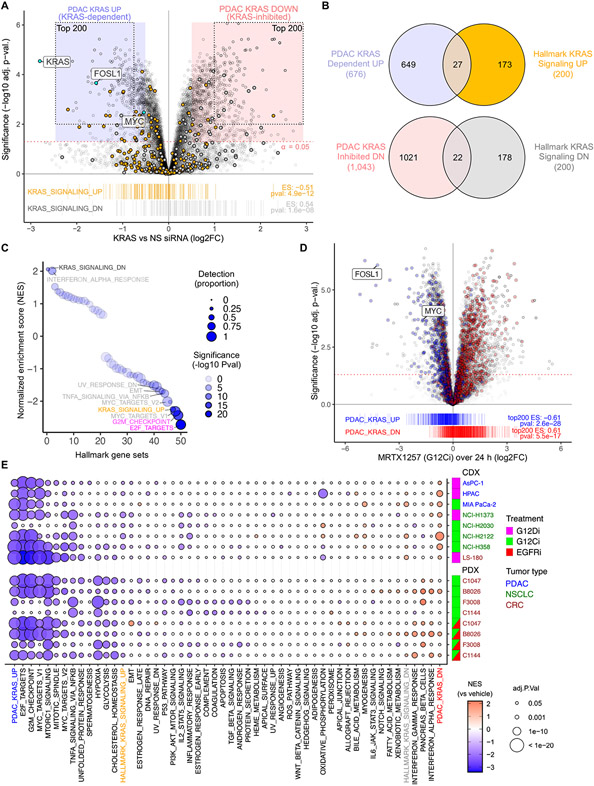
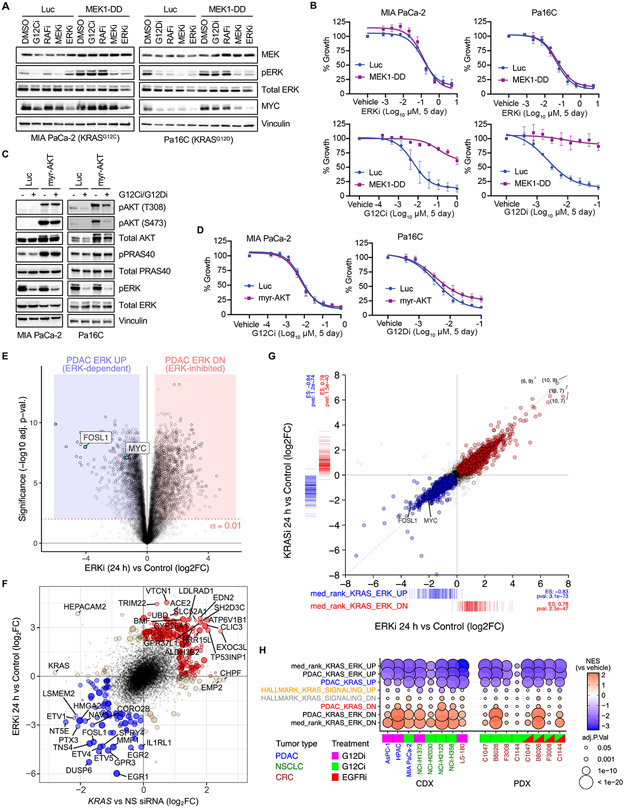
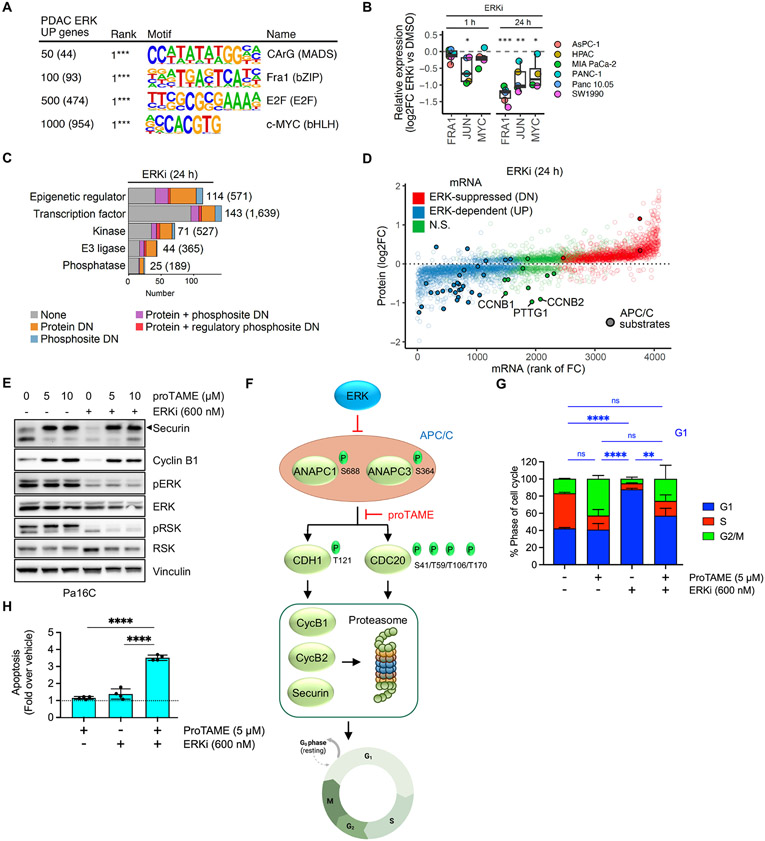
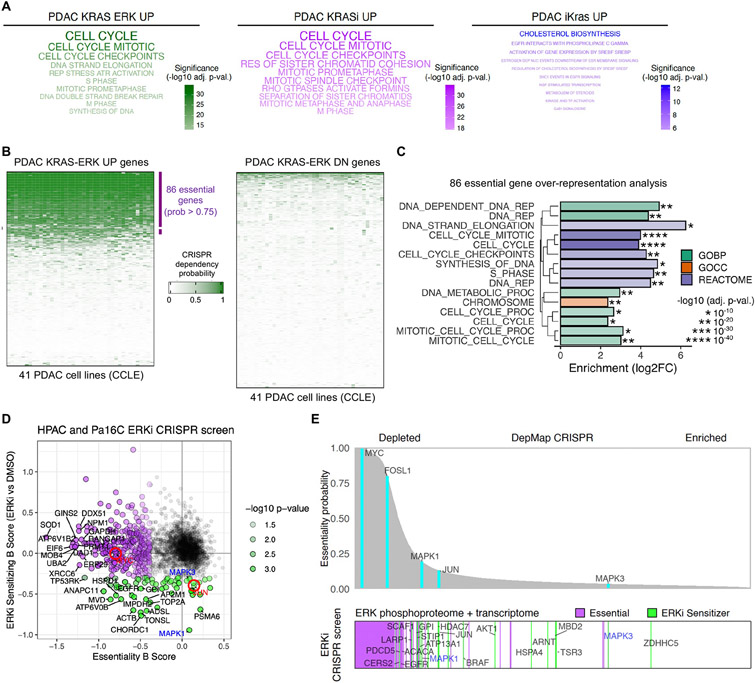
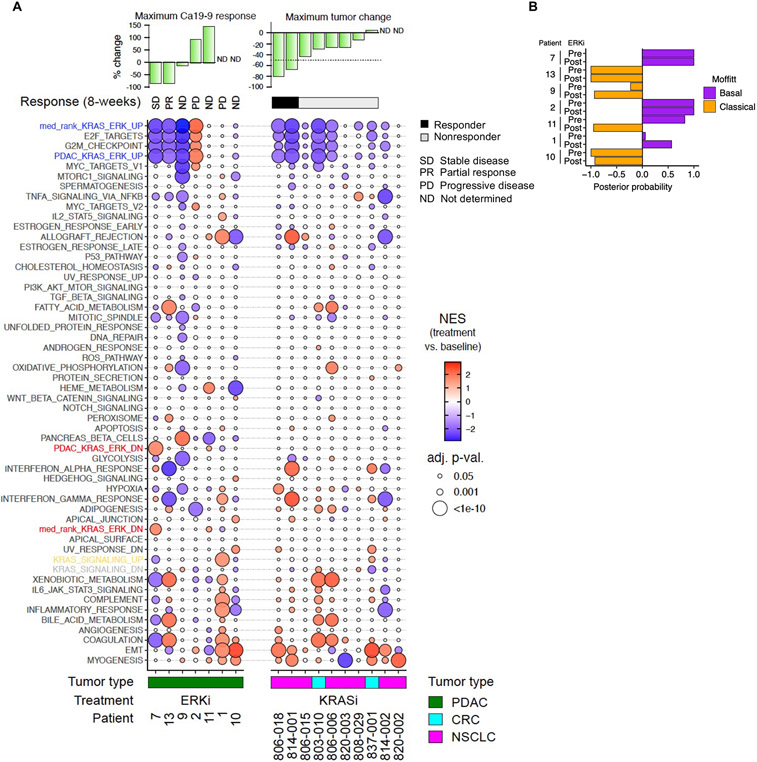
How the KRAS oncogene drives cancer growth remains poorly understood. Therefore, we established a systemwide portrait of KRAS- and extracellular signal-regulated kinase (ERK)-dependent gene transcription in KRAS-mutant cancer to delineate the molecular mechanisms of growth and of inhibitor resistance. Unexpectedly, our KRAS-dependent gene signature diverges substantially from the frequently cited Hallmark KRAS signaling gene signature, is driven predominantly through the ERK mitogen-activated protein kinase (MAPK) cascade, and accurately reflects KRAS- and ERK-regulated gene transcription in KRAS-mutant cancer patients. Integration with our ERK-regulated phospho- and total proteome highlights ERK deregulation of the anaphase promoting complex/cyclosome (APC/C) and other components of the cell cycle machinery as key processes that drive pancreatic ductal adenocarcinoma (PDAC) growth. Our findings elucidate mechanistically the critical role of ERK in driving KRAS-mutant tumor growth and in resistance to KRAS-ERK MAPK targeted therapies.
Figures
References
-
- Hayashi A, Hong J, Iacobuzio-Donahue CA, The pancreatic cancer genome revisited. Nat Rev Gastroenterol Hepatol 18, 469–481 (2021). - PubMed
-
- Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, Marayati R, Kher S, George SD, Xu M, Wang-Gillam A, Samatar AA, Maitra A, Wennerberg K, Petricoin EF, Yin HH, Nelkin B, Cox AD, Yeh JJ, Der CJ, Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell 29, 75–89 (2016). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P50 CA257911/CA/NCI NIH HHS/United States
- T32 CA009156/CA/NCI NIH HHS/United States
- P30 CA016086/CA/NCI NIH HHS/United States
- K99 CA276700/CA/NCI NIH HHS/United States
- T32 GM007040/GM/NIGMS NIH HHS/United States
- U01 CA199235/CA/NCI NIH HHS/United States
- T32 GM119999/GM/NIGMS NIH HHS/United States
- T32 CA071341/CA/NCI NIH HHS/United States
- F32 CA239328/CA/NCI NIH HHS/United States
- R01 GM138520/GM/NIGMS NIH HHS/United States
- R37 CA251877/CA/NCI NIH HHS/United States
- R01 CA042978/CA/NCI NIH HHS/United States
- P01 CA203657/CA/NCI NIH HHS/United States
- R35 CA232113/CA/NCI NIH HHS/United States
- F31 CA275260/CA/NCI NIH HHS/United States
- F32 CA232529/CA/NCI NIH HHS/United States
- P50 CA196510/CA/NCI NIH HHS/United States
- F30 CA243253/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
