

A CD36-dependent non-canonical lipid metabolism program promotes immune escape and resistance to hypomethylating agent therapy in AML
- PMID: 38843841
- PMCID: PMC11228649
- DOI: 10.1016/j.xcrm.2024.101592
A CD36-dependent non-canonical lipid metabolism program promotes immune escape and resistance to hypomethylating agent therapy in AML
Abstract
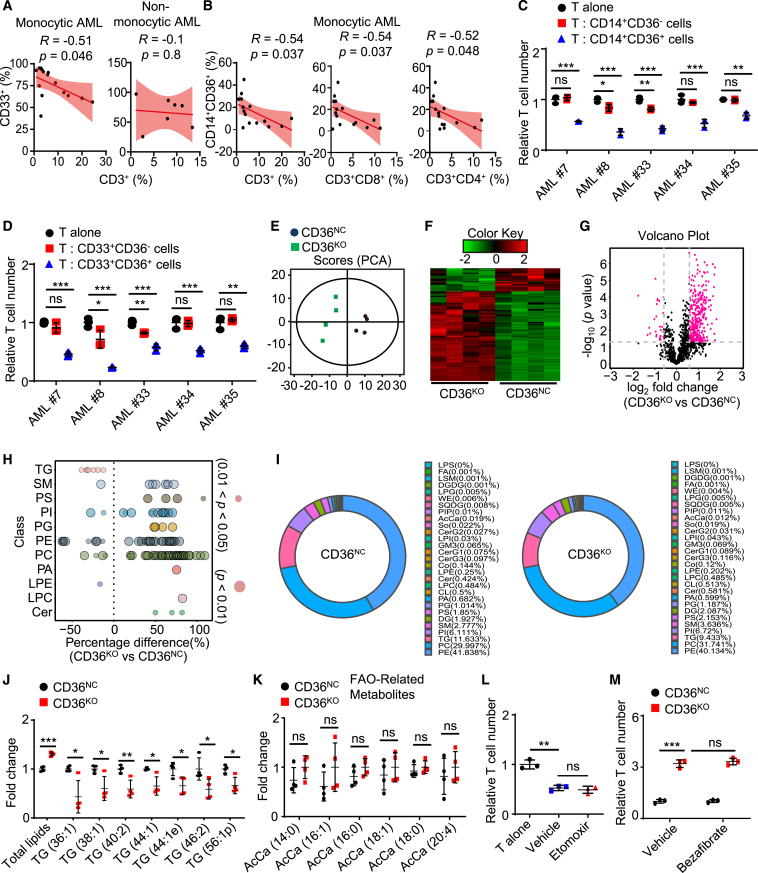
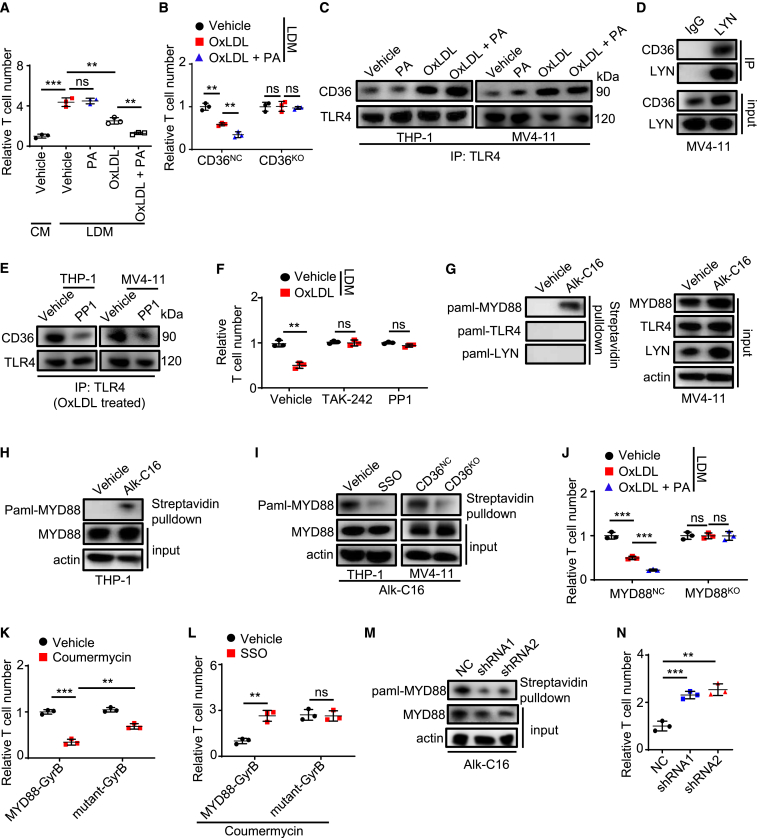
Environmental lipids are essential for fueling tumor energetics, but whether these exogenous lipids transported into cancer cells facilitate immune escape remains unclear. Here, we find that CD36, a transporter for exogenous lipids, promotes acute myeloid leukemia (AML) immune evasion. We show that, separately from its established role in lipid oxidation, CD36 on AML cells senses oxidized low-density lipoprotein (OxLDL) to prime the TLR4-LYN-MYD88-nuclear factor κB (NF-κB) pathway, and exogenous palmitate transfer via CD36 further potentiates this innate immune pathway by supporting ZDHHC6-mediated MYD88 palmitoylation. Subsequently, NF-κB drives the expression of immunosuppressive genes that inhibit anti-tumor T cell responses. Notably, high-fat-diet or hypomethylating agent decitabine treatment boosts the immunosuppressive potential of AML cells by hijacking CD36-dependent innate immune signaling, leading to a dampened therapeutic effect. This work is of translational interest because lipid restriction by US Food and Drug Administration (FDA)-approved lipid-lowering statin drugs improves the efficacy of decitabine therapy by weakening leukemic CD36-mediated immunosuppression.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures






References
-
- Lübbert M., Suciu S., Baila L., Rüter B.H., Platzbecker U., Giagounidis A., Selleslag D., Labar B., Germing U., Salih H.R., et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J. Clin. Oncol. 2011;29:1987–1996. doi: 10.1200/jco.2010.30.9245. - DOI - PubMed
-
- Kantarjian H.M., Thomas X.G., Dmoszynska A., Wierzbowska A., Mazur G., Mayer J., Gau J.P., Chou W.C., Buckstein R., Cermak J., et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J. Clin. Oncol. 2012;30:2670–2677. doi: 10.1200/jco.2011.38.9429. - DOI - PMC - PubMed
-
- Pollyea D.A., Stevens B.M., Jones C.L., Winters A., Pei S., Minhajuddin M., D'Alessandro A., Culp-Hill R., Riemondy K.A., Gillen A.E., et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018;24:1859–1866. doi: 10.1038/s41591-018-0233-1. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
