

C-type natriuretic peptide/cGMP/FoxO3 signaling attenuates hyperproliferation of pericytes from patients with pulmonary arterial hypertension
- PMID: 38844781
- PMCID: PMC11156916
- DOI: 10.1038/s42003-024-06375-3
C-type natriuretic peptide/cGMP/FoxO3 signaling attenuates hyperproliferation of pericytes from patients with pulmonary arterial hypertension
Abstract
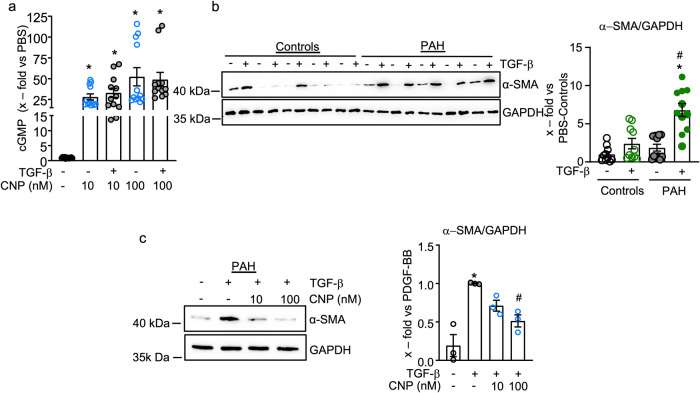
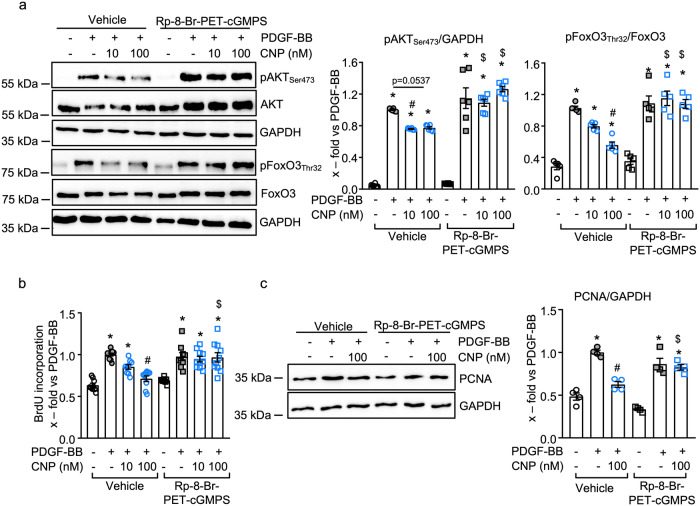
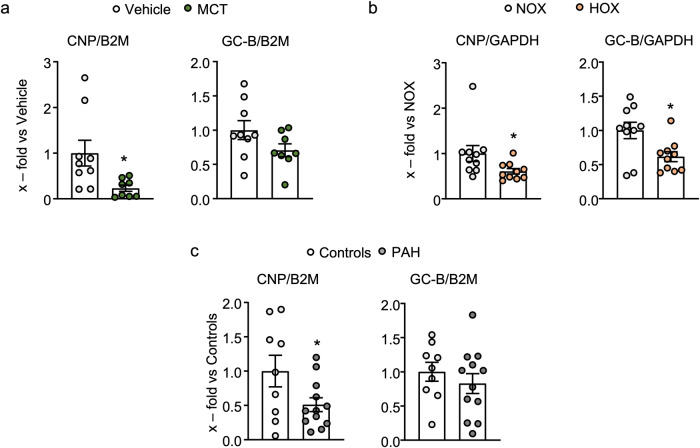
Pericyte dysfunction, with excessive migration, hyperproliferation, and differentiation into smooth muscle-like cells contributes to vascular remodeling in Pulmonary Arterial Hypertension (PAH). Augmented expression and action of growth factors trigger these pathological changes. Endogenous factors opposing such alterations are barely known. Here, we examine whether and how the endothelial hormone C-type natriuretic peptide (CNP), signaling through the cyclic guanosine monophosphate (cGMP) -producing guanylyl cyclase B (GC-B) receptor, attenuates the pericyte dysfunction observed in PAH. The results demonstrate that CNP/GC-B/cGMP signaling is preserved in lung pericytes from patients with PAH and prevents their growth factor-induced proliferation, migration, and transdifferentiation. The anti-proliferative effect of CNP is mediated by cGMP-dependent protein kinase I and inhibition of the Phosphoinositide 3-kinase (PI3K)/AKT pathway, ultimately leading to the nuclear stabilization and activation of the Forkhead Box O 3 (FoxO3) transcription factor. Augmentation of the CNP/GC-B/cGMP/FoxO3 signaling pathway might be a target for novel therapeutics in the field of PAH.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
- DA2462/1-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- KU 1037/8-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- KU 1037/12-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- CRC 1525, 45398101/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 2021_EKEA.131/Else Kröner-Fresenius-Stiftung (Else Kroner-Fresenius Foundation)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
