

DNA methylation patterns of transcription factor binding regions characterize their functional and evolutionary contexts
- PMID: 38844976
- PMCID: PMC11155190
- DOI: 10.1186/s13059-024-03218-6
DNA methylation patterns of transcription factor binding regions characterize their functional and evolutionary contexts
Abstract
Background: DNA methylation is an important epigenetic modification which has numerous roles in modulating genome function. Its levels are spatially correlated across the genome, typically high in repressed regions but low in transcription factor (TF) binding sites and active regulatory regions. However, the mechanisms establishing genome-wide and TF binding site methylation patterns are still unclear.
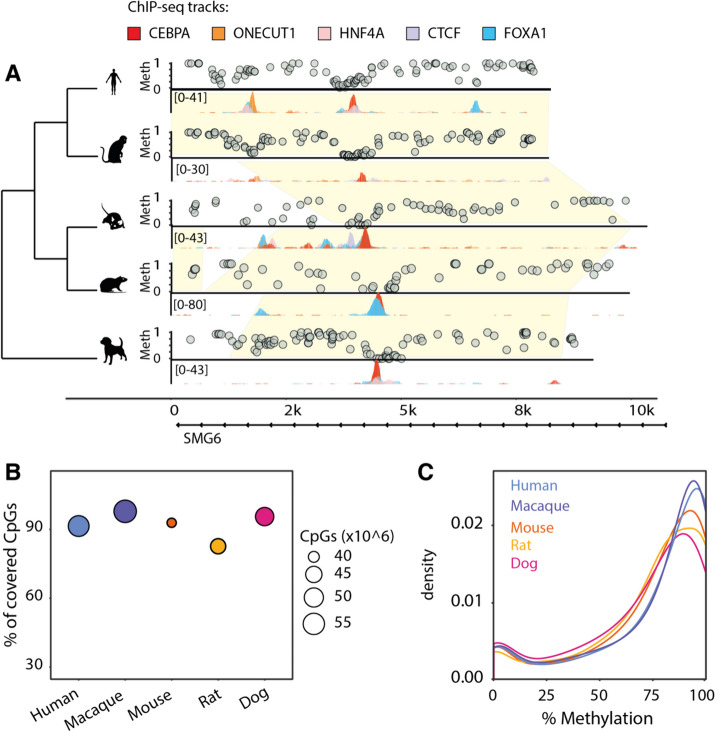
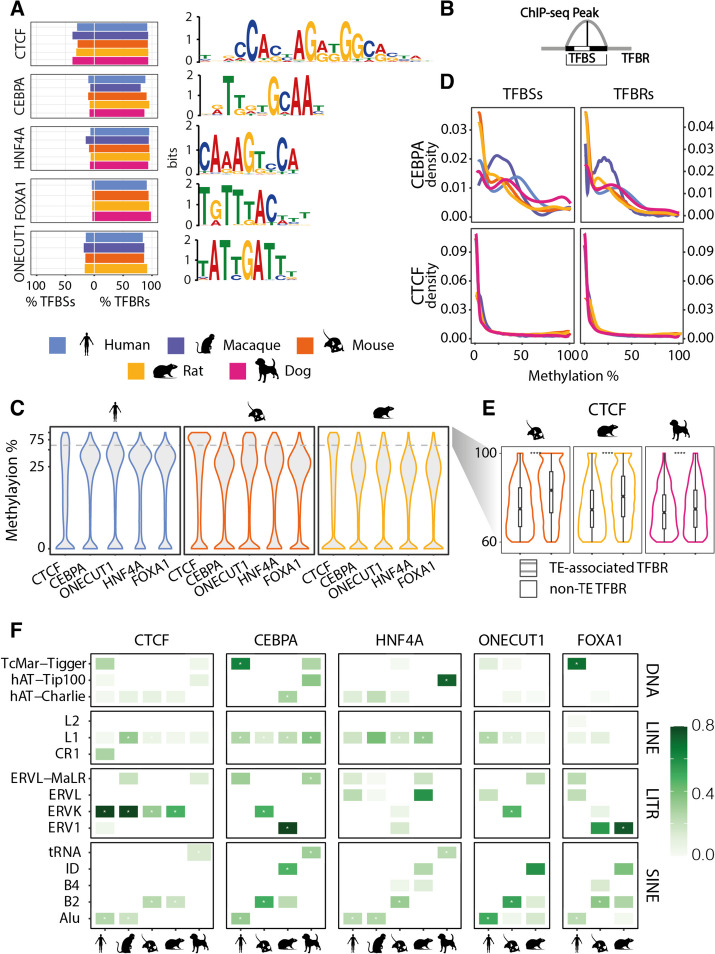
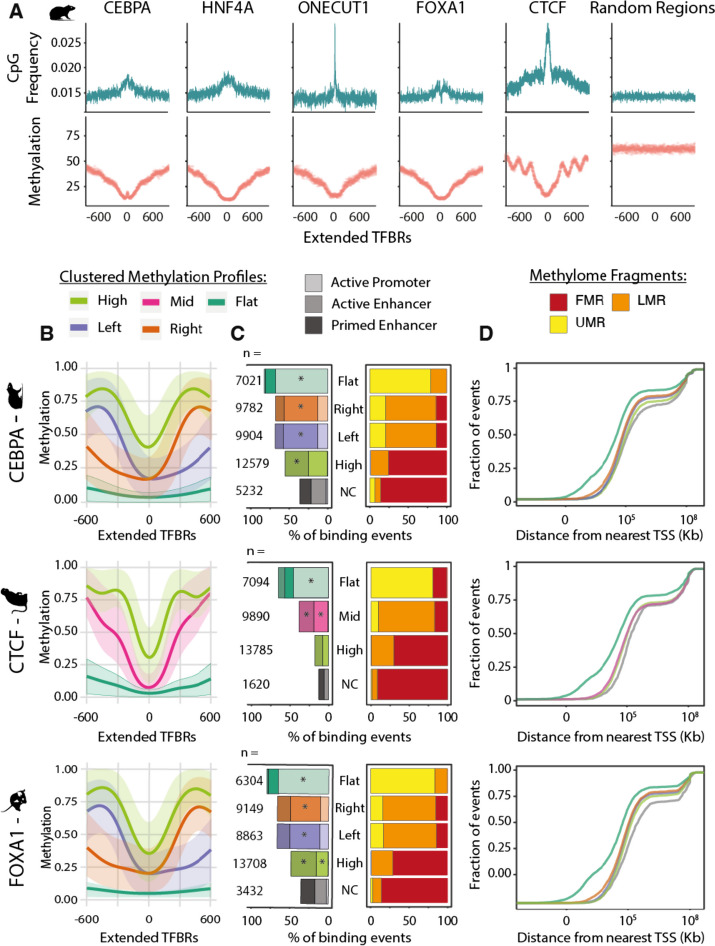
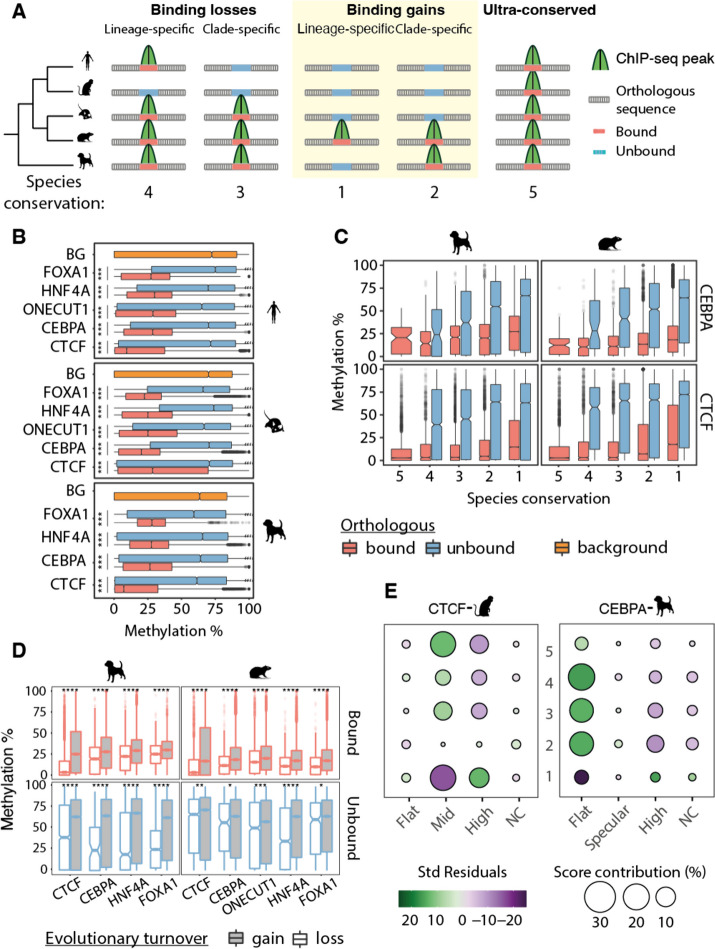
Results: Here we use a comparative approach to investigate the association of DNA methylation to TF binding evolution in mammals. Specifically, we experimentally profile DNA methylation and combine this with published occupancy profiles of five distinct TFs (CTCF, CEBPA, HNF4A, ONECUT1, FOXA1) in the liver of five mammalian species (human, macaque, mouse, rat, dog). TF binding sites are lowly methylated, but they often also have intermediate methylation levels. Furthermore, biding sites are influenced by the methylation status of CpGs in their wider binding regions even when CpGs are absent from the core binding motif. Employing a classification and clustering approach, we extract distinct and species-conserved patterns of DNA methylation levels at TF binding regions. CEBPA, HNF4A, ONECUT1, and FOXA1 share the same methylation patterns, while CTCF's differ. These patterns characterize alternative functions and chromatin landscapes of TF-bound regions. Leveraging our phylogenetic framework, we find DNA methylation gain upon evolutionary loss of TF occupancy, indicating coordinated evolution. Furthermore, each methylation pattern has its own evolutionary trajectory reflecting its genomic contexts.
Conclusions: Our epigenomic analyses indicate a role for DNA methylation in TF binding changes across species including that specific DNA methylation profiles characterize TF binding and are associated with their regulatory activity, chromatin contexts, and evolutionary trajectories.
Keywords: DNA methylation; Evolution; Mammals; Transcription factor binding.
© 2024. The Author(s).
Conflict of interest statement
P.F. is a member of the Scientific Advisory Boards of Fabric Genomics, Inc., and Eagle Genomics, Ltd. All other authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
