

Photoswitching of Co(ii)-based coordination cages containing azobenzene backbones
- PMID: 38846406
- PMCID: PMC11151815
- DOI: 10.1039/d4sc01575d
Photoswitching of Co(ii)-based coordination cages containing azobenzene backbones
Abstract
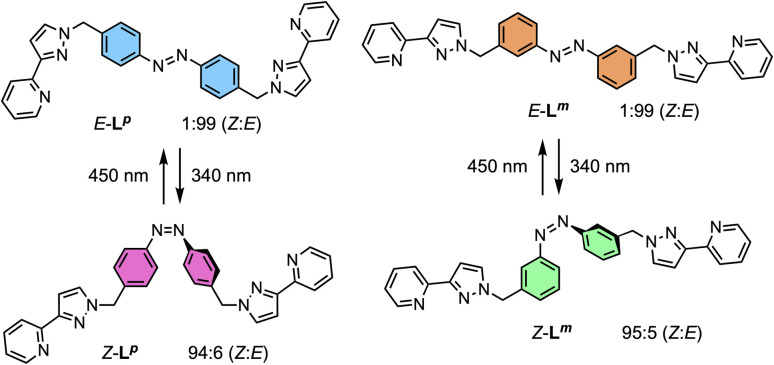
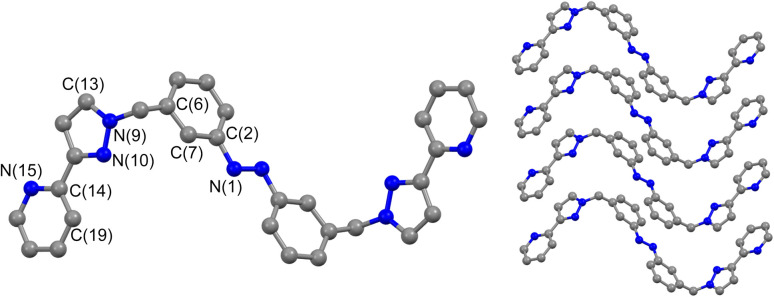
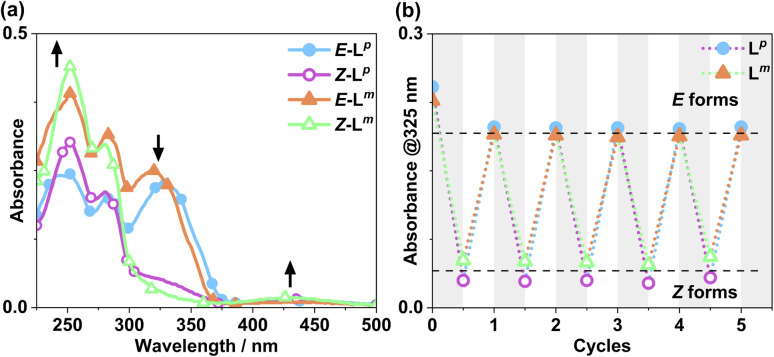
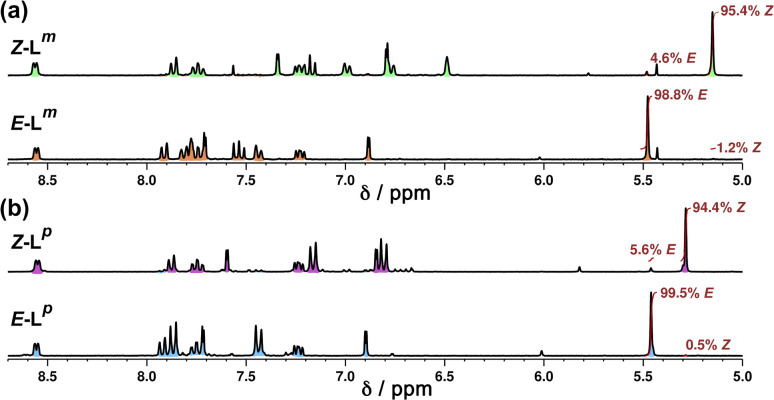
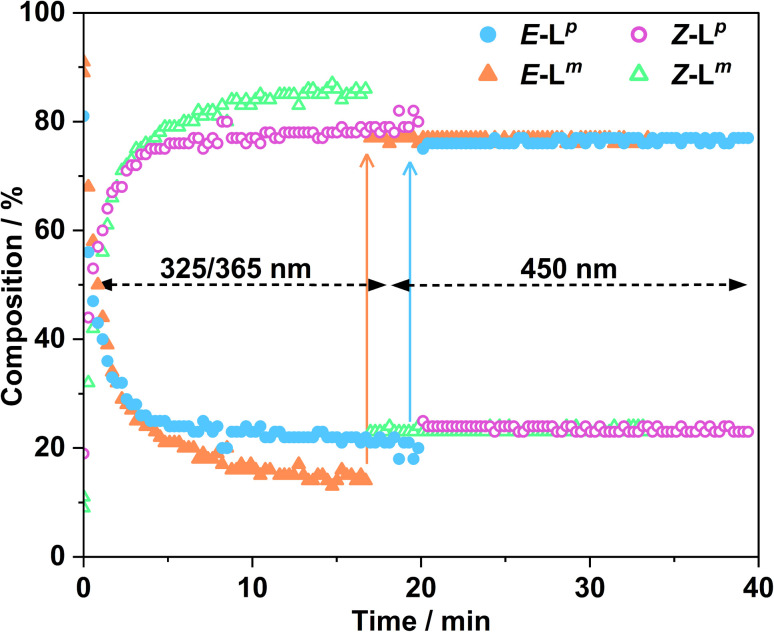
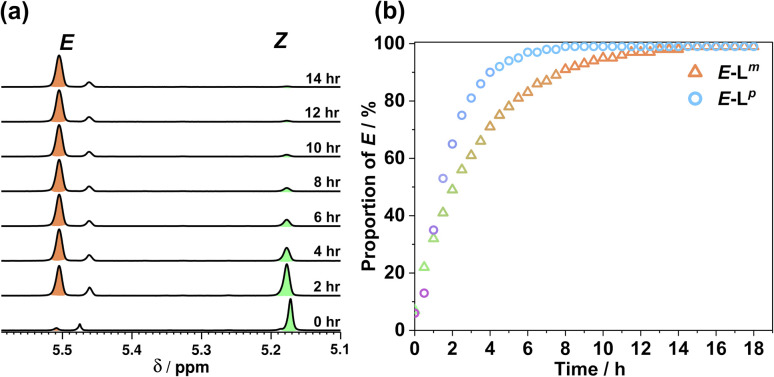
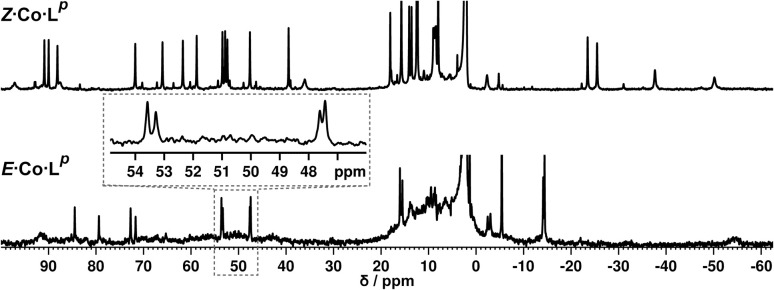
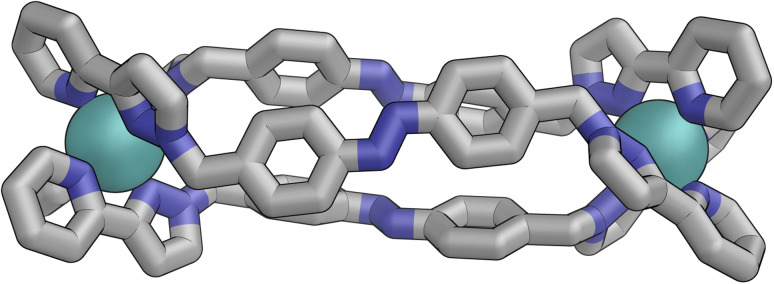
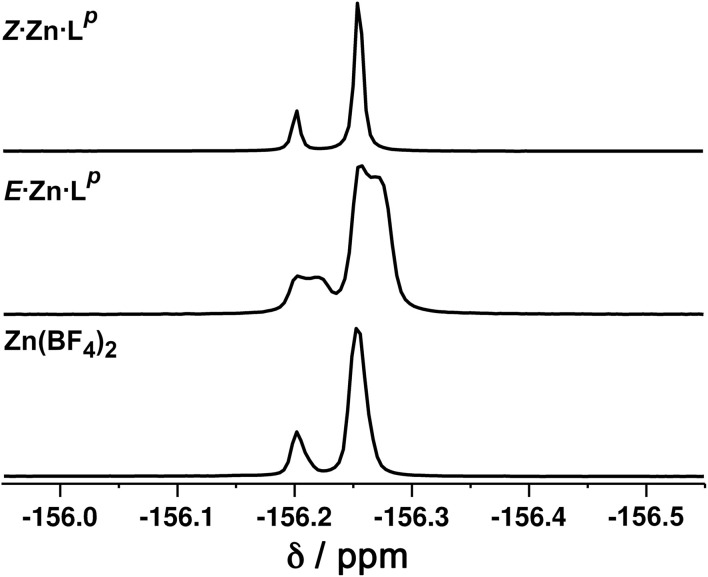
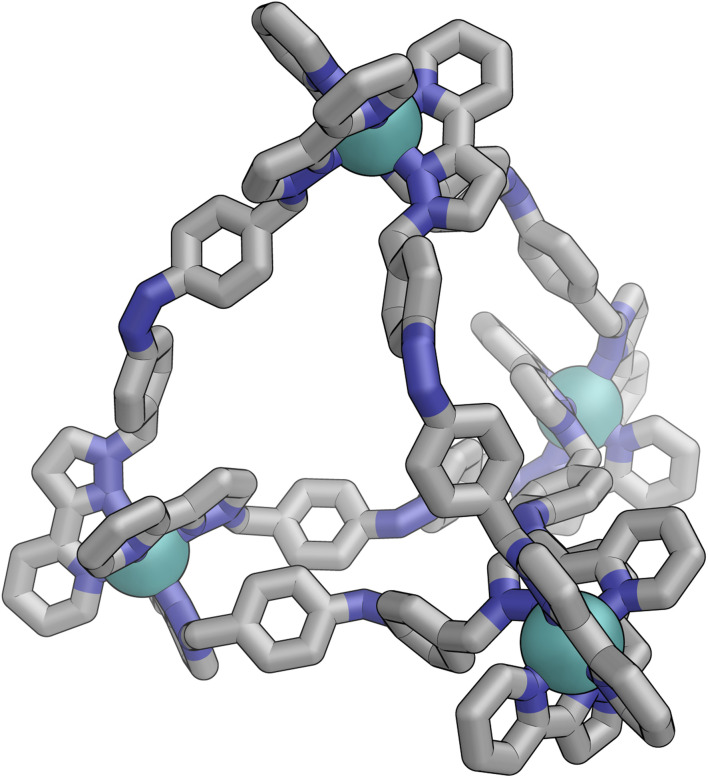
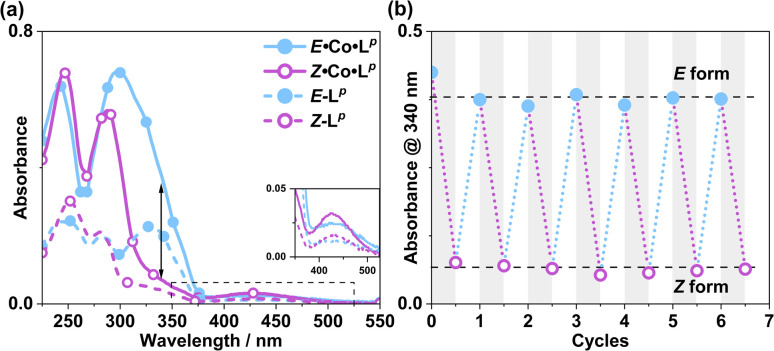
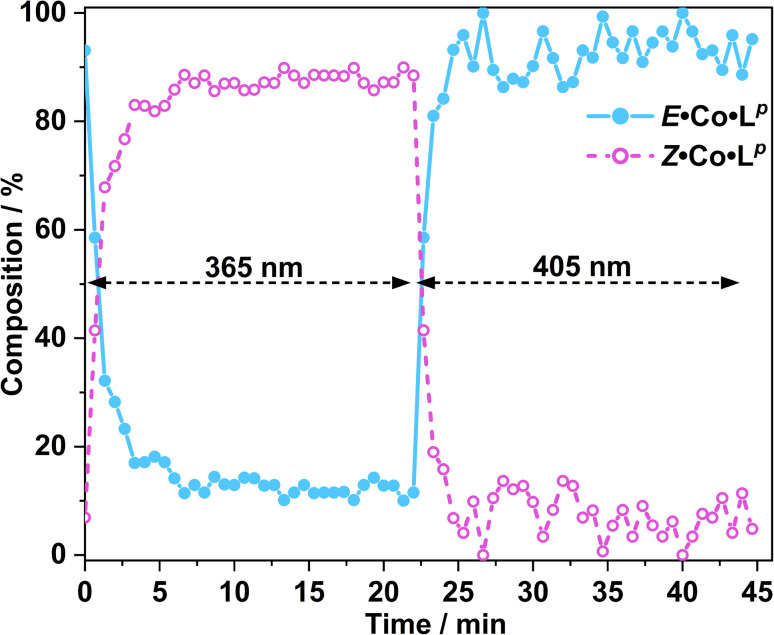
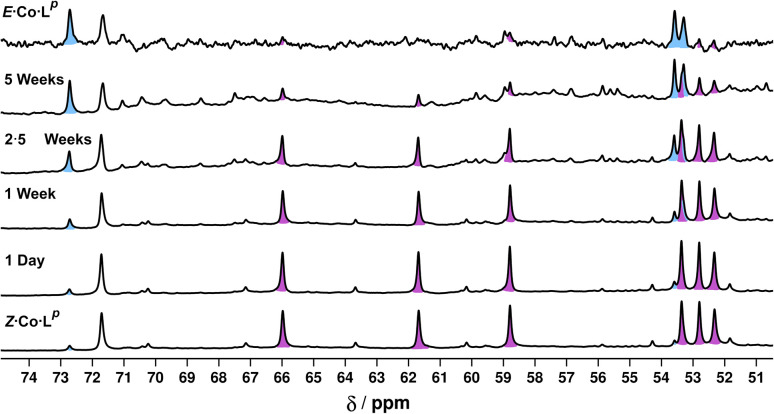
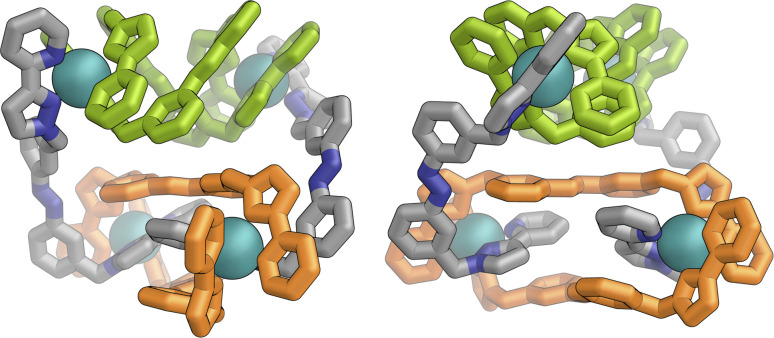
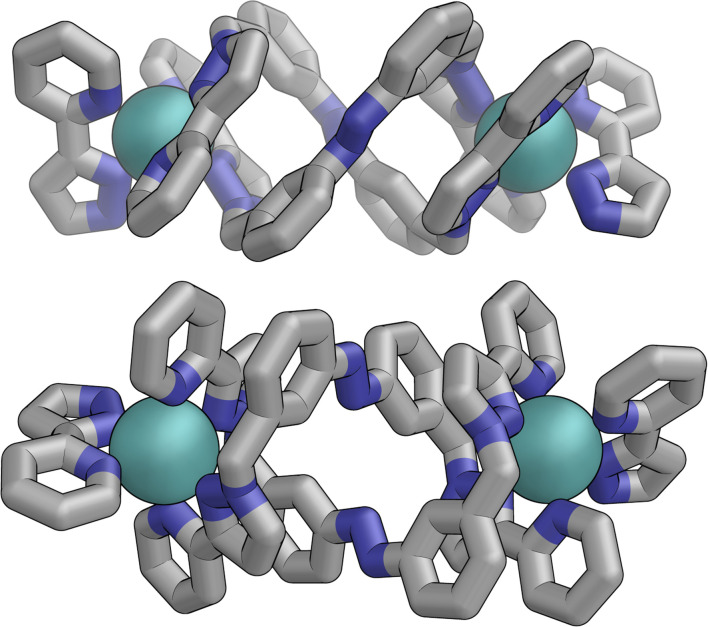
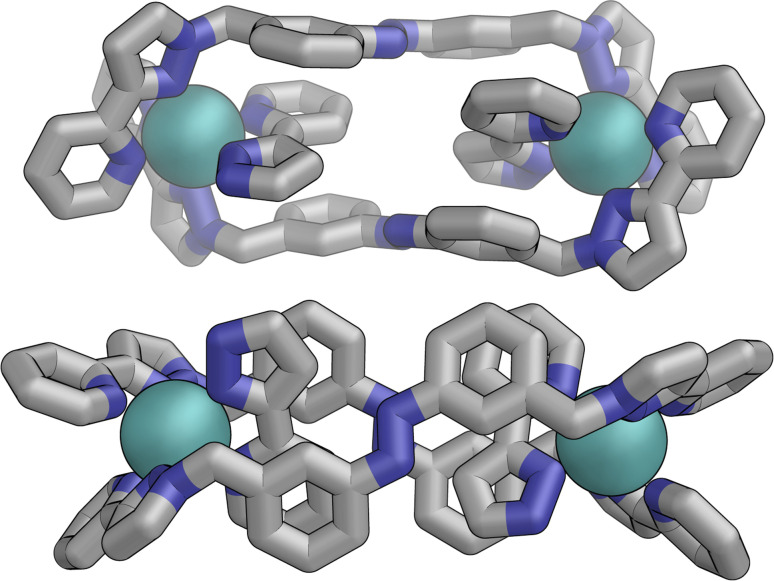
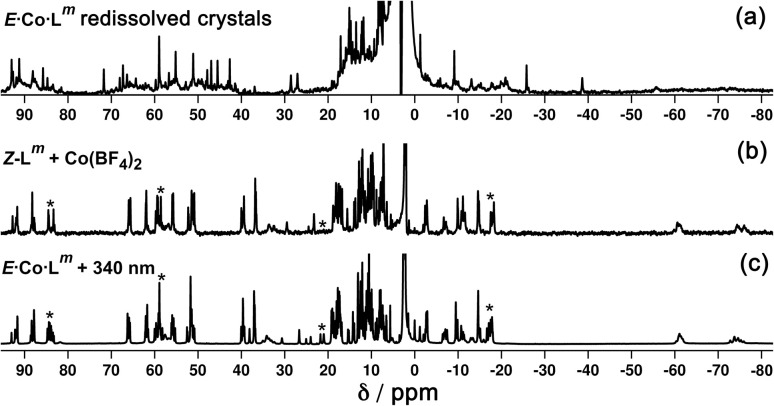
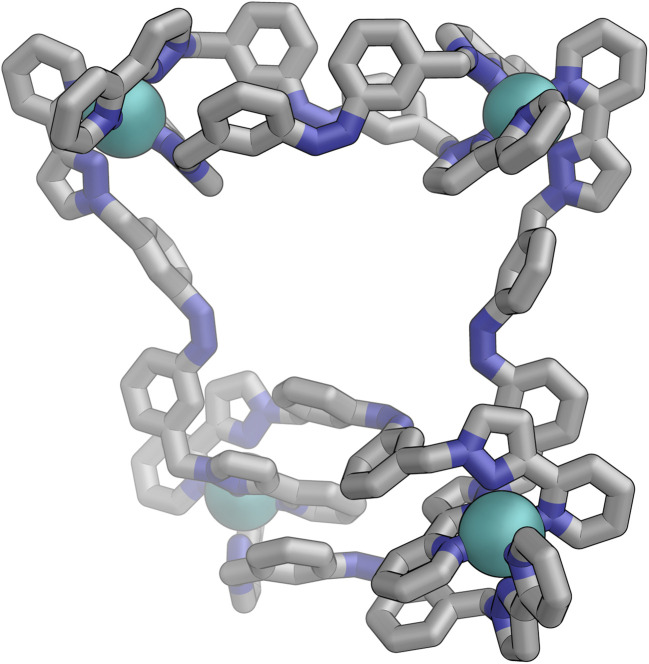
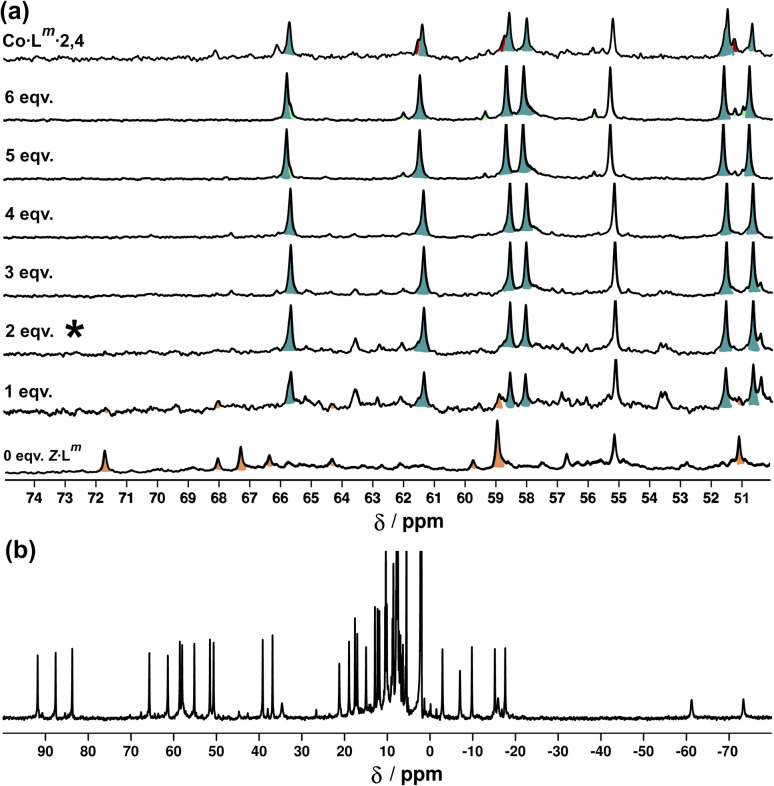
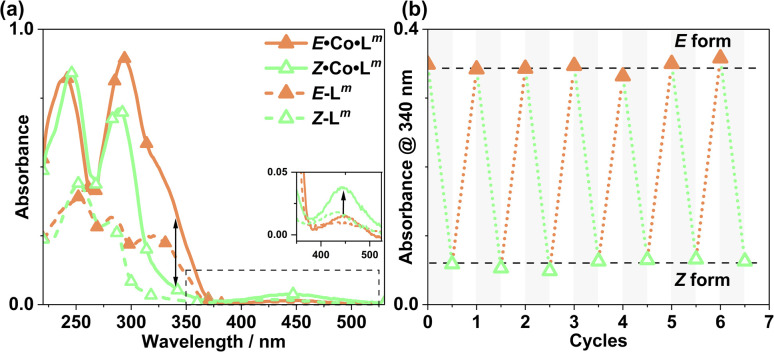
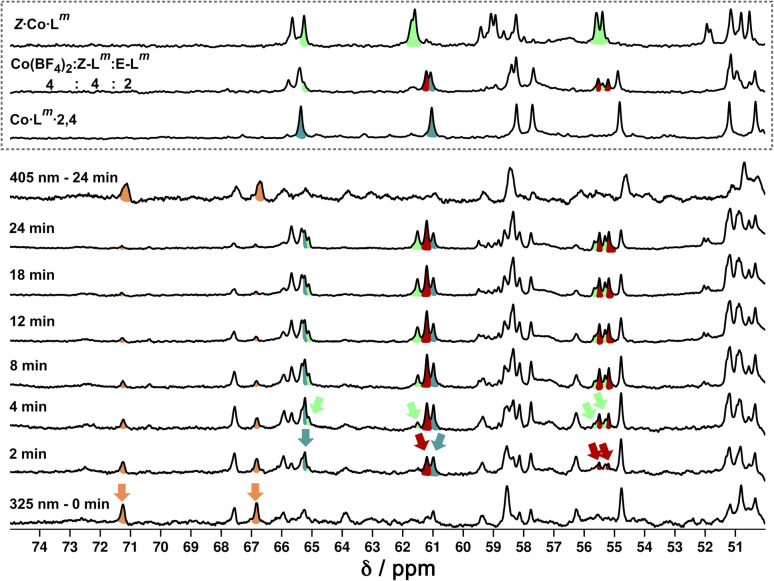
Inclusion of photoswitchable azobenzene units as spacers into ditopic bridging ligands Lm and Lp, containing two chelating pyrazolyl-pyridine termini, allows formation of metal complex assemblies with Co(ii) that undergo a range of light-induced structural transformations. One notable result is the light-induced conversion of a Co2(Lp)3 dinuclear triple helicate (based on the E ligand isomer) to a C 3-symmetric Co4(Lp)6 assembly, assumed to be an edge-bridged tetrahedral cage, based on the Z ligand isomer. Another is the preparation of a series of Co4(Lm)6 complexes, of which Co4(E-Lm)6 was crystallographically characterised and consists of a pair of Co2(Lm)2 double helicates connected by an additional two bridging ligands which span the pair of helicate units, giving a cyclic Co4 array in which one and then two bridging ligands alternate around the periphery. A set of Co4(Lm)6 complexes could be prepared containing different ratios of Z : E ligand isomers (0 : 6, 2 : 4, 4 : 2 and 6 : 0) of which Co4(Z-Lm)2(E-Lm)4 was particularly stable and dominated the speciation behaviour, either during light-induced switching of the ligand geometry in pre-formed complexes, or when ligand isomers were combined in different proportions during the preparation. These examples of (i) interconversion between Co2L3 (helicate) and (ii) Co4L6 (cage) assemblies with Lp, and the interconversion between a series of Co4L6 assemblies Co4(Z-Lm)n(E-Lm)6-n with Lm, constitute significant advances in the field of photoswitchable supramolecular assemblies.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures
Similar articles
-
An interconverting family of coordination cages and a meso-helicate; effects of temperature, concentration, and solvent on the product distribution of a self-assembly process.Inorg Chem. 2015 Mar 16;54(6):2626-37. doi: 10.1021/ic502780b. Epub 2015 Feb 20. Inorg Chem. 2015. PMID: 25700155
-
Coordination chemistry of tetradentate N-donor ligands containing two pyrazolyl-pyridine units separated by a 1,8-naphthyl spacer: dodecanuclear and tetranuclear coordination cages and cyclic helicates.Inorg Chem. 2006 May 15;45(10):3905-19. doi: 10.1021/ic060157d. Inorg Chem. 2006. PMID: 16676949
-
Mixed-ligand molecular paneling: dodecanuclear cuboctahedral coordination cages based on a combination of edge-bridging and face-capping ligands.J Am Chem Soc. 2008 Sep 3;130(35):11641-9. doi: 10.1021/ja803847w. Epub 2008 Aug 8. J Am Chem Soc. 2008. PMID: 18686952
-
Molecular squares, cubes and chains from self-assembly of bis-bidentate bridging ligands with transition metal dications.Dalton Trans. 2011 Oct 28;40(40):10360-9. doi: 10.1039/c1dt10263j. Epub 2011 Apr 26. Dalton Trans. 2011. PMID: 21519621
-
Palladium(II)-Based Self-Assembled Heteroleptic Coordination Architectures: A Growing Family.Chemistry. 2019 Sep 20;25(53):12241-12269. doi: 10.1002/chem.201900831. Epub 2019 Jul 12. Chemistry. 2019. PMID: 31158303 Review.
References
-
- Cook T. R. Stang P. J. Chem. Rev. 2015;115:7001. doi: 10.1021/cr5005666. - DOI - PubMed
- Smulders M. M. J. Riddell I. A. Browne C. Nitschke J. R. Chem. Soc. Rev. 2013;42:1728. doi: 10.1039/C2CS35254K. - DOI - PubMed
- Zhang D. Ronson T. K. Nitschke J. R. Acc. Chem. Res. 2018;51:2423. doi: 10.1021/acs.accounts.8b00303. - DOI - PubMed
- Vardhan H. Yusubov M. Verpoort F. Coord. Chem. Rev. 2016;306:171. doi: 10.1016/j.ccr.2015.05.016. - DOI
- Percástegui E. G. Ronson T. K. Nitschke J. R. Chem. Rev. 2020;120:13480. doi: 10.1021/acs.chemrev.0c00672. - DOI - PMC - PubMed
- Rizzuto F. J. von Krbek L. K. S. Nitschke J. R. Nat. Rev. Chem. 2019;3:204. doi: 10.1038/s41570-019-0085-3. - DOI
- Ronson T. K. Meng W. Nitschke J. R. J. Am. Chem. Soc. 2017;139:9698. doi: 10.1021/jacs.7b05202. - DOI - PubMed
- Taylor C. G. P. Argent S. P. Ludden M. D. Piper J. R. Mozaceanu C. Barnett S. A. Ward M. D. Chem.–Eur. J. 2020;26:3054. doi: 10.1002/chem.201905499. - DOI - PMC - PubMed
- Cullen W. Turega S. Hunter C. A. Ward M. D. Chem. Sci. 2015;6:2790. doi: 10.1039/C5SC00534E. - DOI - PMC - PubMed
-
- Qu D.-H. Wang Q.-C. Zhang Q.-W. Ma X. Tian H. Chem. Rev. 2015;115:7543–7588. doi: 10.1021/cr5006342. - DOI - PubMed
- Wezenberg S. J. Chem. Commun. 2022;58:11045. doi: 10.1039/D2CC04329G. - DOI - PMC - PubMed
- Ghosh A. Slappendel L. Nguyen B.-N. T. von Krbek L. K. S. Ronson T. K. Castilla A. M. Nitschke J. R. J. Am. Chem. Soc. 2023;145:3828. doi: 10.1021/jacs.2c10084. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous