

Accessing the electronic structure of liquid crystalline semiconductors with bottom-up electronic coarse-graining
- PMID: 38846409
- PMCID: PMC11151863
- DOI: 10.1039/d3sc06749a
Accessing the electronic structure of liquid crystalline semiconductors with bottom-up electronic coarse-graining
Abstract
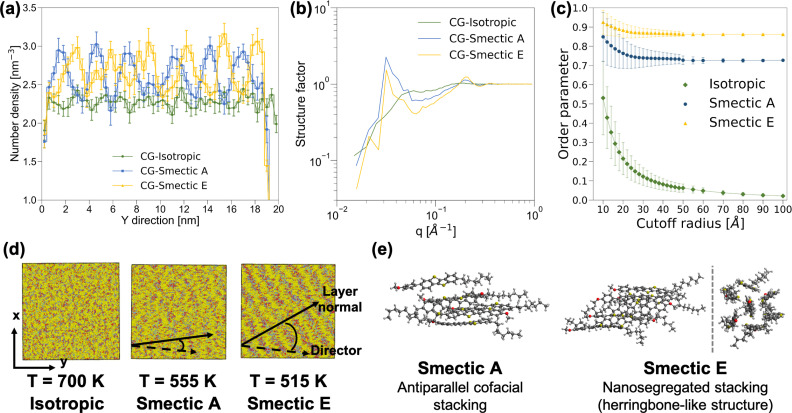
Understanding the relationship between multiscale morphology and electronic structure is a grand challenge for semiconducting soft materials. Computational studies aimed at characterizing these relationships require the complex integration of quantum-chemical (QC) calculations, all-atom and coarse-grained (CG) molecular dynamics simulations, and back-mapping approaches. However, these methods pose substantial computational challenges that limit their application to the requisite length scales of soft material morphologies. Here, we demonstrate the bottom-up electronic coarse-graining (ECG) of morphology-dependent electronic structure in the liquid-crystal-forming semiconductor, 2-(4-methoxyphenyl)-7-octyl-benzothienobenzothiophene (BTBT). ECG is applied to construct density functional theory (DFT)-accurate valence band Hamiltonians of the isotropic and smectic liquid crystal (LC) phases using only the CG representation of BTBT. By bypassing the atomistic resolution and its prohibitive computational costs, ECG enables the first calculations of the morphology dependence of the electronic structure of charge carriers across LC phases at the ∼20 nm length scale, with robust statistical sampling. Kinetic Monte Carlo (kMC) simulations reveal a strong morphology dependence on zero-field charge mobility among different LC phases as well as the presence of two-molecule charge carriers that act as traps and hinder charge transport. We leverage these results to further evaluate the feasibility of developing mesoscopic, field-based ECG models in future works. The fully CG approach to electronic property predictions in LC semiconductors opens a new computational direction for designing electronic processes in soft materials at their characteristic length scales.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures






References
-
- Luo D. Jang W. Babu D. D. Kim M. S. Wang D. H. Kyaw A. K. K. J. Mater. Chem. A. 2022;10:3255–3295. doi: 10.1039/D1TA10707K. - DOI
-
- Bernardo G. Lopes T. Lidzey D. G. Mendes A. Adv. Energy Mater. 2021;11:2100342. doi: 10.1002/aenm.202100342. - DOI
-
- Liu K. Ouyang B. Guo X. Guo Y. Liu Y. npj Flexible Electron. 2022;6:1–19. doi: 10.1038/s41528-022-00133-3. - DOI
LinkOut - more resources
Full Text Sources