

Redox regulation of UPR signalling and mitochondrial ER contact sites
- PMID: 38847861
- PMCID: PMC11335286
- DOI: 10.1007/s00018-024-05286-0
Redox regulation of UPR signalling and mitochondrial ER contact sites
Abstract
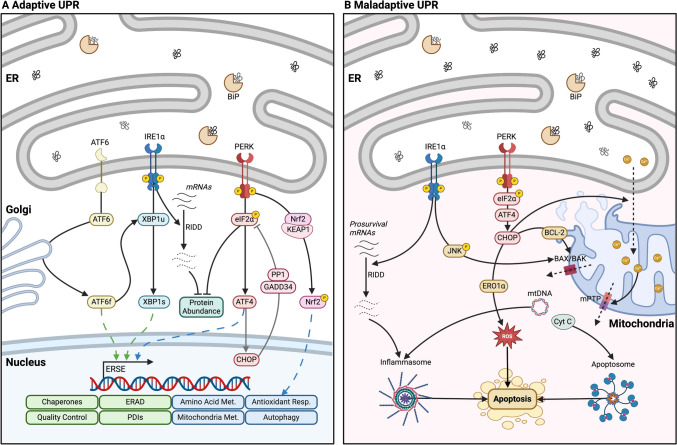
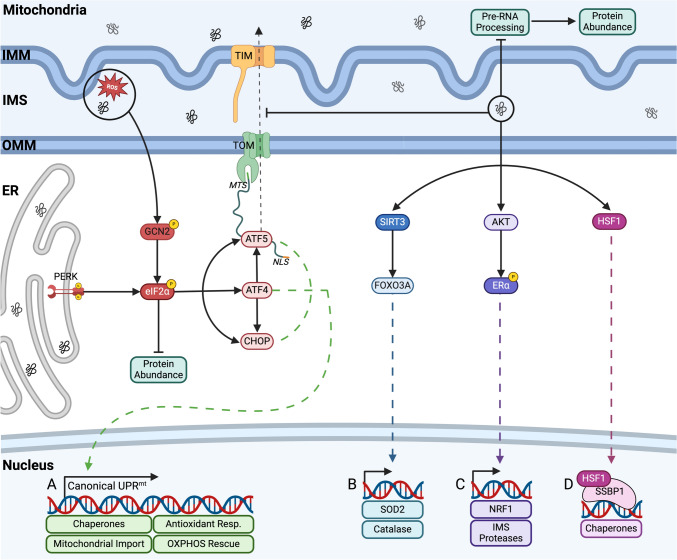
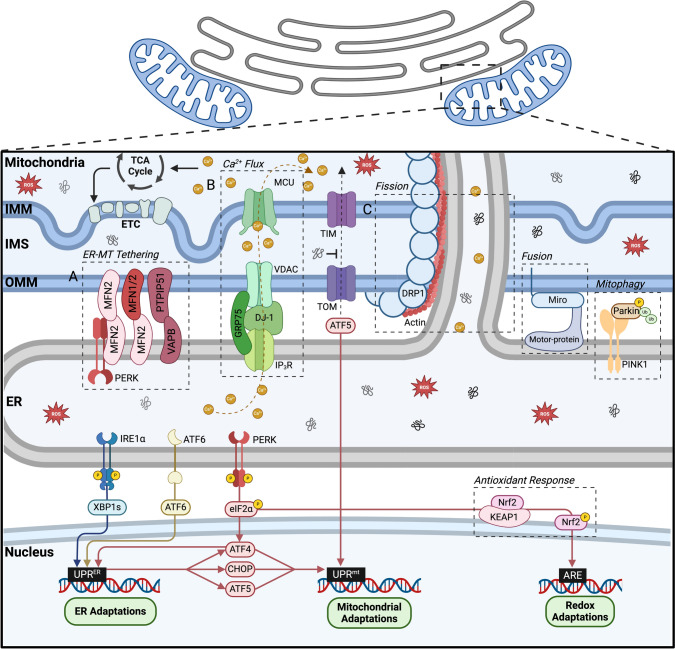
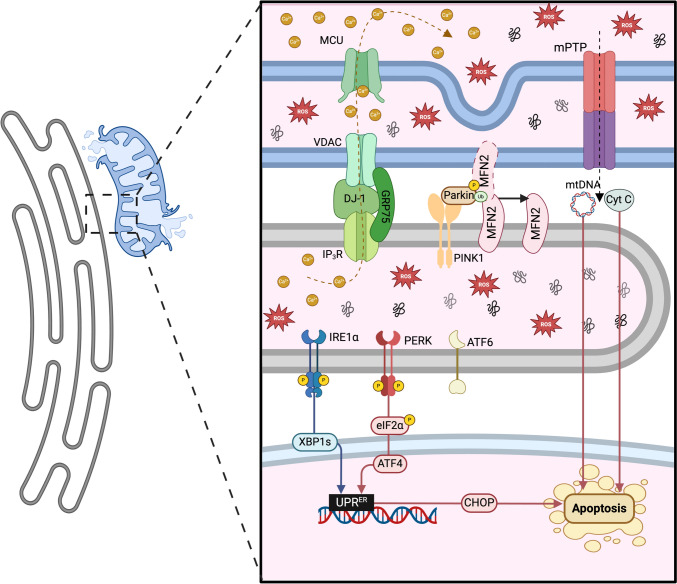
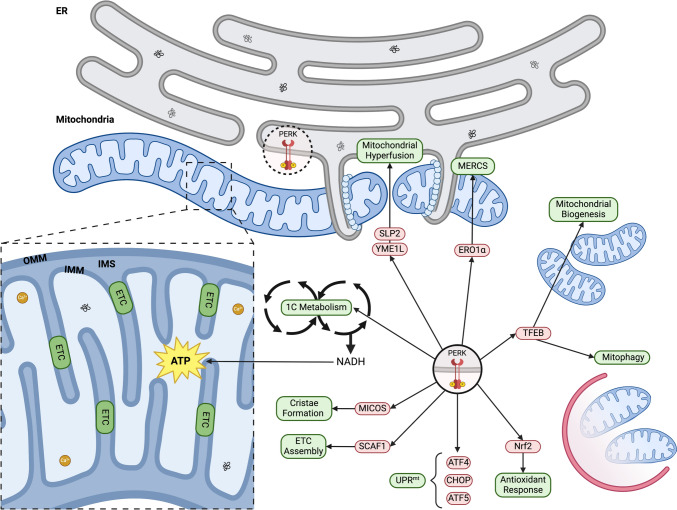
Mitochondria and the endoplasmic reticulum (ER) have a synergistic relationship and are key regulatory hubs in maintaining cell homeostasis. Communication between these organelles is mediated by mitochondria ER contact sites (MERCS), allowing the exchange of material and information, modulating calcium homeostasis, redox signalling, lipid transfer and the regulation of mitochondrial dynamics. MERCS are dynamic structures that allow cells to respond to changes in the intracellular environment under normal homeostatic conditions, while their assembly/disassembly are affected by pathophysiological conditions such as ageing and disease. Disruption of protein folding in the ER lumen can activate the Unfolded Protein Response (UPR), promoting the remodelling of ER membranes and MERCS formation. The UPR stress receptor kinases PERK and IRE1, are located at or close to MERCS. UPR signalling can be adaptive or maladaptive, depending on whether the disruption in protein folding or ER stress is transient or sustained. Adaptive UPR signalling via MERCS can increase mitochondrial calcium import, metabolism and dynamics, while maladaptive UPR signalling can result in excessive calcium import and activation of apoptotic pathways. Targeting UPR signalling and the assembly of MERCS is an attractive therapeutic approach for a range of age-related conditions such as neurodegeneration and sarcopenia. This review highlights the emerging evidence related to the role of redox mediated UPR activation in orchestrating inter-organelle communication between the ER and mitochondria, and ultimately the determination of cell function and fate.
Keywords: C. elegans; Contact-sites; Hormesis; Mitochondrial dynamics; Redox signalling; Skeletal muscle.
© 2024. The Author(s).
Conflict of interest statement
Not applicable.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
