

An eQTL-based approach reveals candidate regulators of LINE-1 RNA levels in lymphoblastoid cells
- PMID: 38848448
- PMCID: PMC11189215
- DOI: 10.1371/journal.pgen.1011311
An eQTL-based approach reveals candidate regulators of LINE-1 RNA levels in lymphoblastoid cells
Abstract
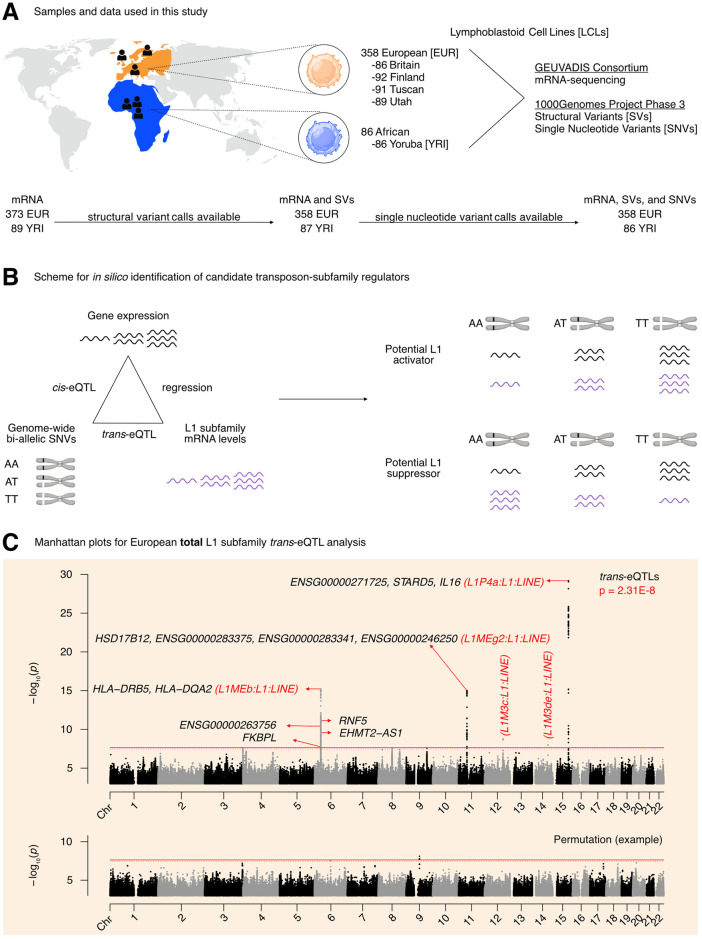
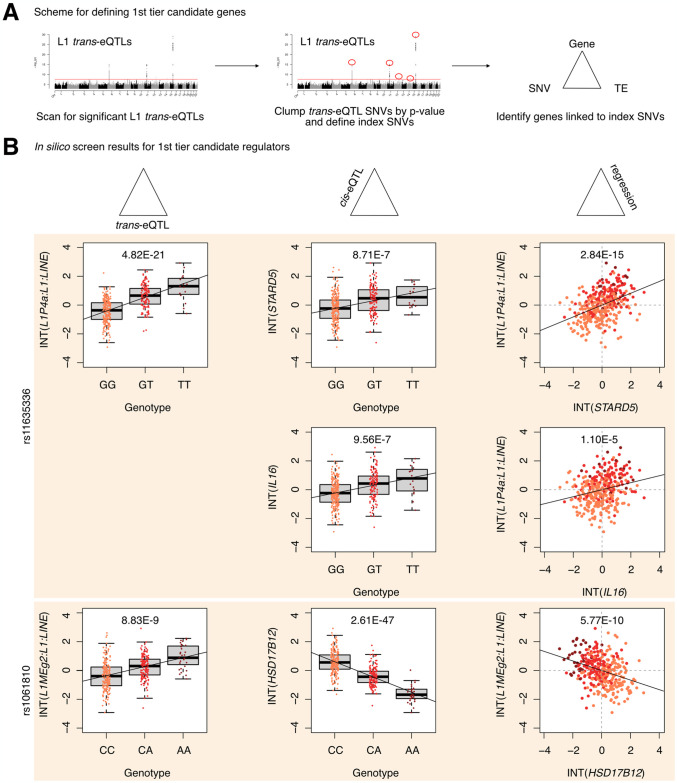
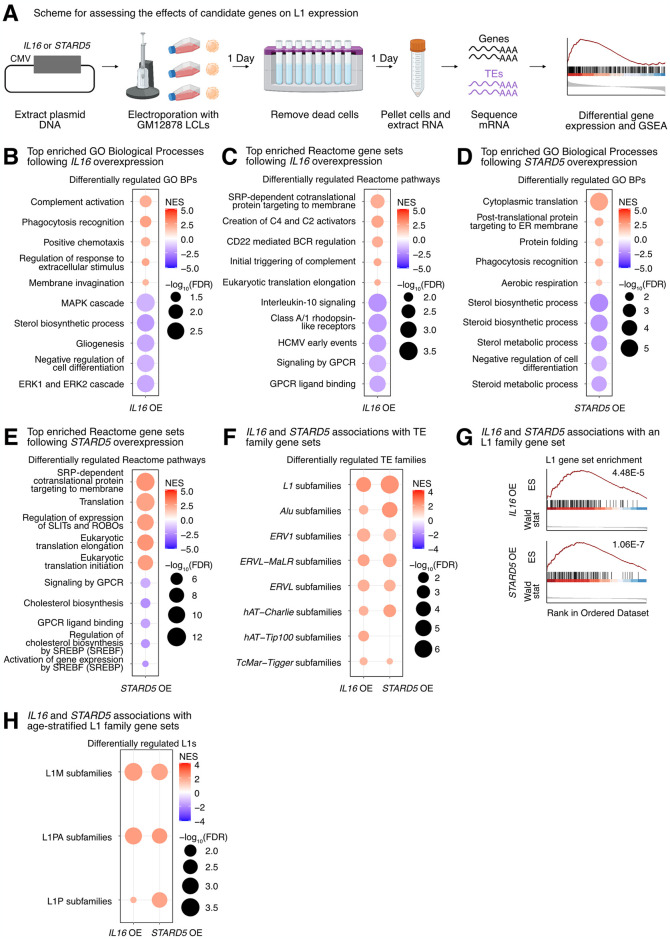
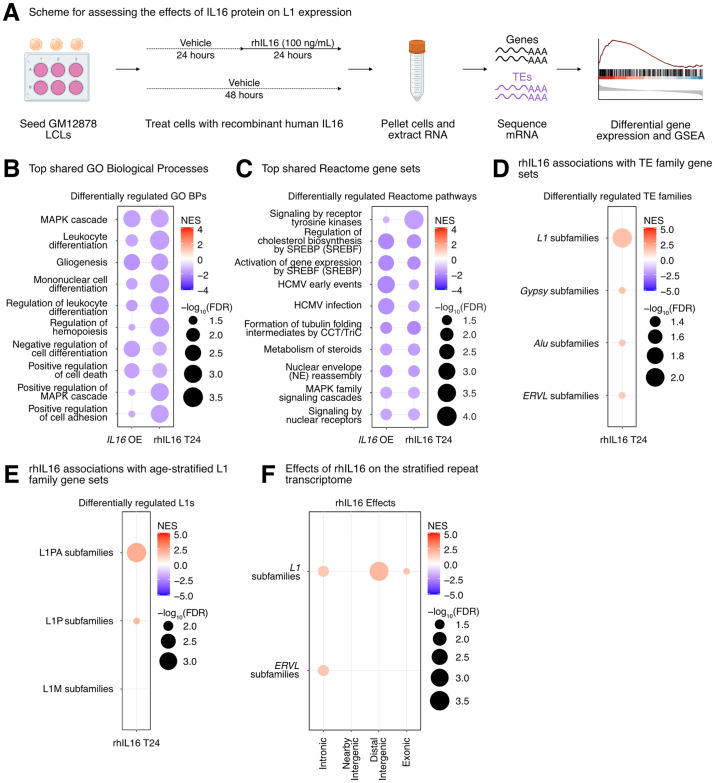
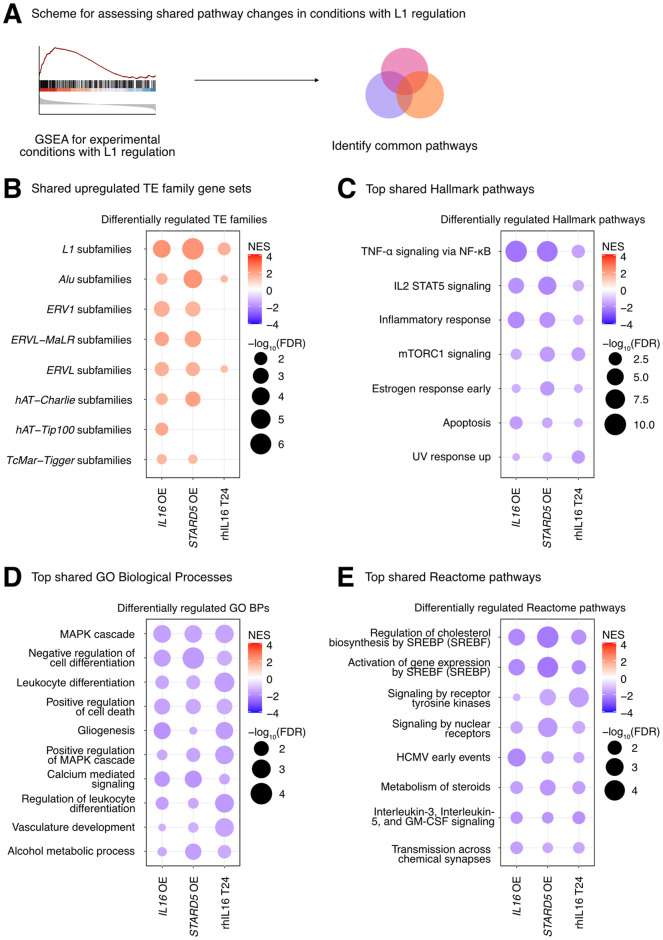
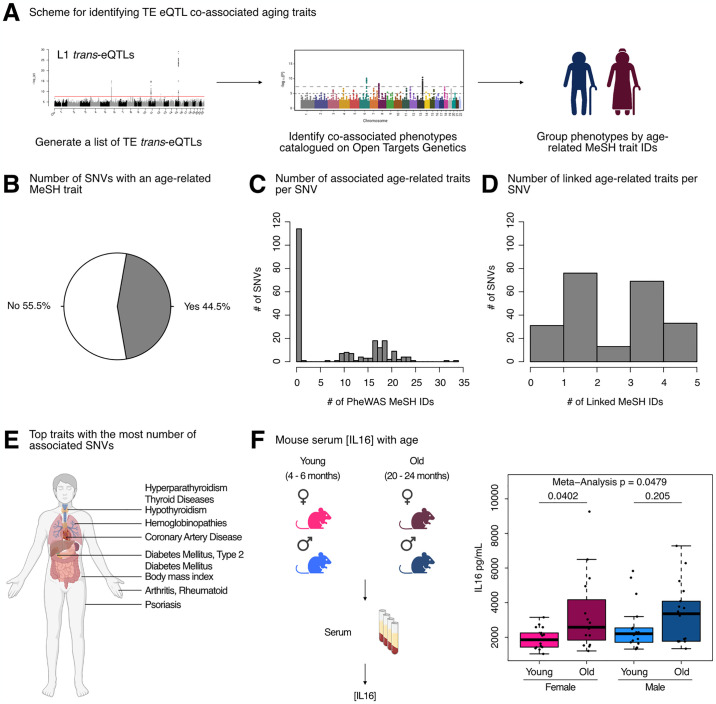
Long interspersed element 1 (LINE-1; L1) are a family of transposons that occupy ~17% of the human genome. Though a small number of L1 copies remain capable of autonomous transposition, the overwhelming majority of copies are degenerate and immobile. Nevertheless, both mobile and immobile L1s can exert pleiotropic effects (promoting genome instability, inflammation, or cellular senescence) on their hosts, and L1's contributions to aging and aging diseases is an area of active research. However, because of the cell type-specific nature of transposon control, the catalogue of L1 regulators remains incomplete. Here, we employ an eQTL approach leveraging transcriptomic and genomic data from the GEUVADIS and 1000Genomes projects to computationally identify new candidate regulators of L1 RNA levels in lymphoblastoid cell lines. To cement the role of candidate genes in L1 regulation, we experimentally modulate the levels of top candidates in vitro, including IL16, STARD5, HSD17B12, and RNF5, and assess changes in TE family expression by Gene Set Enrichment Analysis (GSEA). Remarkably, we observe subtle but widespread upregulation of TE family expression following IL16 and STARD5 overexpression. Moreover, a short-term 24-hour exposure to recombinant human IL16 was sufficient to transiently induce subtle, but widespread, upregulation of L1 subfamilies. Finally, we find that many L1 expression-associated genetic variants are co-associated with aging traits across genome-wide association study databases. Our results expand the catalogue of genes implicated in L1 RNA control and further suggest that L1-derived RNA contributes to aging processes. Given the ever-increasing availability of paired genomic and transcriptomic data, we anticipate this new approach to be a starting point for more comprehensive computational scans for regulators of transposon RNA levels.
Copyright: © 2024 Bravo et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Update of
-
An eQTL-based Approach Reveals Candidate Regulators of LINE-1 RNA Levels in Lymphoblastoid Cells.bioRxiv [Preprint]. 2023 Nov 29:2023.08.15.553416. doi: 10.1101/2023.08.15.553416. bioRxiv. 2023. Update in: PLoS Genet. 2024 Jun 7;20(6):e1011311. doi: 10.1371/journal.pgen.1011311. PMID: 37645920 Free PMC article. Updated. Preprint.
Similar articles
-
An eQTL-based Approach Reveals Candidate Regulators of LINE-1 RNA Levels in Lymphoblastoid Cells.bioRxiv [Preprint]. 2023 Nov 29:2023.08.15.553416. doi: 10.1101/2023.08.15.553416. bioRxiv. 2023. Update in: PLoS Genet. 2024 Jun 7;20(6):e1011311. doi: 10.1371/journal.pgen.1011311. PMID: 37645920 Free PMC article. Updated. Preprint.
-
RNA Next-Generation Sequencing and a Bioinformatics Pipeline to Identify Expressed LINE-1s at the Locus-Specific Level.J Vis Exp. 2019 May 19;(147):10.3791/59771. doi: 10.3791/59771. J Vis Exp. 2019. PMID: 31157783 Free PMC article.
-
miR-128 Restriction of LINE-1 (L1) Retrotransposition Is Dependent on Targeting hnRNPA1 mRNA.Int J Mol Sci. 2019 Apr 21;20(8):1955. doi: 10.3390/ijms20081955. Int J Mol Sci. 2019. PMID: 31010097 Free PMC article.
-
Long interspersed element-1 (LINE-1): passenger or driver in human neoplasms?PLoS Genet. 2013 Mar;9(3):e1003402. doi: 10.1371/journal.pgen.1003402. Epub 2013 Mar 28. PLoS Genet. 2013. PMID: 23555307 Free PMC article. Review.
-
The human L1 element: a potential biomarker in cancer prognosis, current status and future directions.Curr Mol Med. 2011 Jun;11(4):286-303. doi: 10.2174/156652411795677954. Curr Mol Med. 2011. PMID: 21506922 Review.
Cited by
-
SVA Regulation of Transposable Element Clustered Transcription within the Major Histocompatibility Complex Genomic Class II Region of the Parkinson's Progression Markers Initiative.Genes (Basel). 2024 Sep 9;15(9):1185. doi: 10.3390/genes15091185. Genes (Basel). 2024. PMID: 39336776 Free PMC article.
-
Multi-ancestry GWAS reveals loci linked to human variation in LINE-1- and Alu-insertion numbers.Transl Med Aging. 2025;9:25-40. doi: 10.1016/j.tma.2025.02.001. Epub 2025 Feb 13. Transl Med Aging. 2025. PMID: 40051556 Free PMC article.
-
Comparative Analysis of Transposable Element Evolution in Crustaceans.Genome Biol Evol. 2025 Jul 3;17(7):evaf115. doi: 10.1093/gbe/evaf115. Genome Biol Evol. 2025. PMID: 40462399 Free PMC article.
-
A multi-omics analysis of human fibroblasts overexpressing an Alu transposon reveals widespread disruptions in aging-associated pathways.bioRxiv [Preprint]. 2025 Jul 17:2025.07.11.664466. doi: 10.1101/2025.07.11.664466. bioRxiv. 2025. PMID: 40791480 Free PMC article. Preprint.
-
Multi-ancestry GWAS reveals loci linked to human variation in LINE-1- and Alu-insertion numbers.bioRxiv [Preprint]. 2025 Jan 27:2024.09.10.612283. doi: 10.1101/2024.09.10.612283. bioRxiv. 2025. Update in: Transl Med Aging. 2025;9:25-40. doi: 10.1016/j.tma.2025.02.001. PMID: 39314493 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
