

This is a preprint.
diaTracer enables spectrum-centric analysis of diaPASEF proteomics data
- PMID: 38854051
- PMCID: PMC11160675
- DOI: 10.1101/2024.05.25.595875
diaTracer enables spectrum-centric analysis of diaPASEF proteomics data
Update in
-
diaTracer enables spectrum-centric analysis of diaPASEF proteomics data.Nat Commun. 2025 Jan 2;16(1):95. doi: 10.1038/s41467-024-55448-8. Nat Commun. 2025. PMID: 39747075 Free PMC article.
Abstract
Data-independent acquisition (DIA) has become a widely used strategy for peptide and protein quantification in mass spectrometry-based proteomics studies. The integration of ion mobility separation into DIA analysis, such as the diaPASEF technology available on Bruker's timsTOF platform, further improves the quantification accuracy and protein depth achievable using DIA. We introduce diaTracer, a new spectrum-centric computational tool optimized for diaPASEF data. diaTracer performs three-dimensional (m/z, retention time, ion mobility) peak tracing and feature detection to generate precursor-resolved "pseudo-MS/MS" spectra, facilitating direct ("spectral-library free") peptide identification and quantification from diaPASEF data. diaTracer is available as a stand-alone tool and is fully integrated into the widely used FragPipe computational platform. We demonstrate the performance of diaTracer and FragPipe using diaPASEF data from triple-negative breast cancer (TNBC), cerebrospinal fluid (CSF), and plasma samples, data from phosphoproteomics and HLA immunopeptidomics experiments, and low-input data from a spatial proteomics study. We also show that diaTracer enables unrestricted identification of post-translational modifications from diaPASEF data using open/mass-offset searches.
Conflict of interest statement
Competing interests A.I.N. and F.Y. receive royalties from the University of Michigan for the sale of MSFragger and IonQuant software licenses to commercial entities. All license transactions are managed by the University of Michigan Innovation Partnerships office, and all proceeds are subject to university technology transfer policy. Other authors declare no other competing interests.
Figures

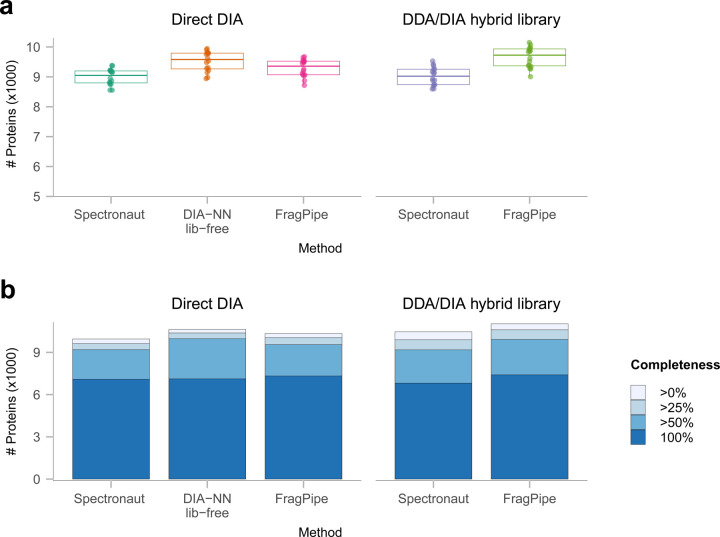





Similar articles
-
diaTracer enables spectrum-centric analysis of diaPASEF proteomics data.Nat Commun. 2025 Jan 2;16(1):95. doi: 10.1038/s41467-024-55448-8. Nat Commun. 2025. PMID: 39747075 Free PMC article.
-
Evaluation of DDA Library-Free Strategies for Phosphoproteomics and Ubiquitinomics Data-Independent Acquisition Data.J Proteome Res. 2023 Jul 7;22(7):2232-2245. doi: 10.1021/acs.jproteome.2c00735. Epub 2023 May 31. J Proteome Res. 2023. PMID: 37256709
-
diaPASEF Analysis for HLA-I Peptides Enables Quantification of Common Cancer Neoantigens.Mol Cell Proteomics. 2025 Apr;24(4):100938. doi: 10.1016/j.mcpro.2025.100938. Epub 2025 Mar 3. Mol Cell Proteomics. 2025. PMID: 40044040 Free PMC article.
-
Data-Independent Acquisition Mass Spectrometry-Based Proteomics and Software Tools: A Glimpse in 2020.Proteomics. 2020 Sep;20(17-18):e1900276. doi: 10.1002/pmic.201900276. Epub 2020 May 19. Proteomics. 2020. PMID: 32275110 Review.
-
Deep learning approaches for data-independent acquisition proteomics.Expert Rev Proteomics. 2021 Dec;18(12):1031-1043. doi: 10.1080/14789450.2021.2020654. Epub 2021 Dec 28. Expert Rev Proteomics. 2021. PMID: 34918987 Review.
References
-
- Aebersold R. & Mann M. Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355 (2016). - PubMed
-
- Venable J.D., Dong M.-Q., Wohlschlegel J., Dillin A. & Yates J.R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nature Methods 1, 39–45 (2004). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials