

LINC00982-encoded protein PRDM16-DT regulates CHEK2 splicing to suppress colorectal cancer metastasis and chemoresistance
- PMID: 38855188
- PMCID: PMC11155395
- DOI: 10.7150/thno.95485
LINC00982-encoded protein PRDM16-DT regulates CHEK2 splicing to suppress colorectal cancer metastasis and chemoresistance
Abstract
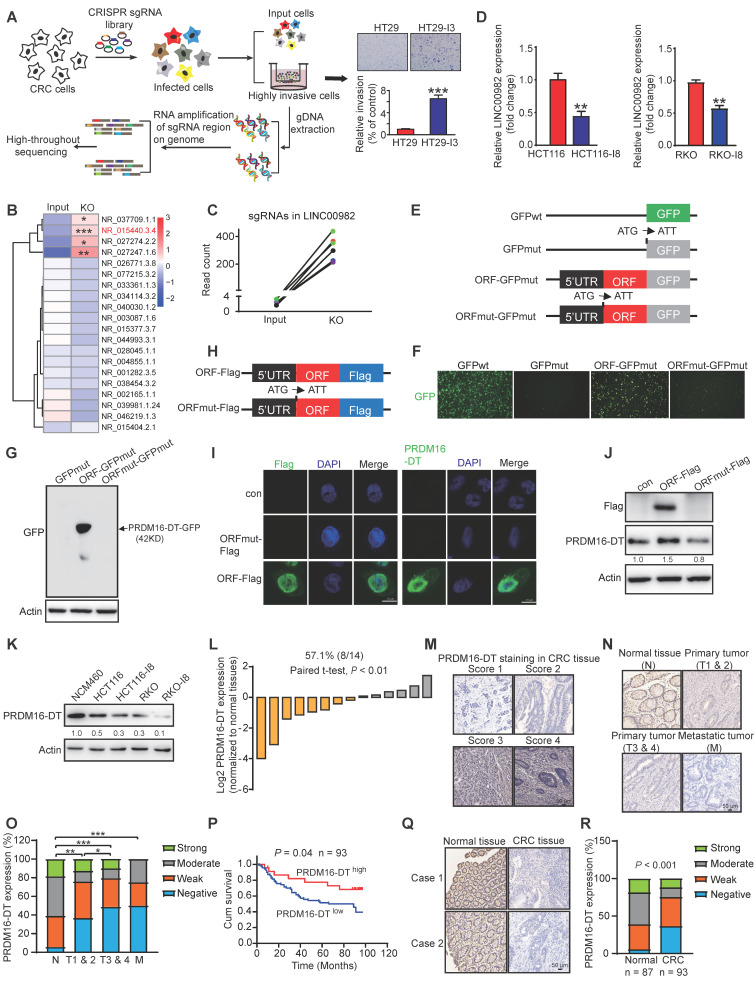
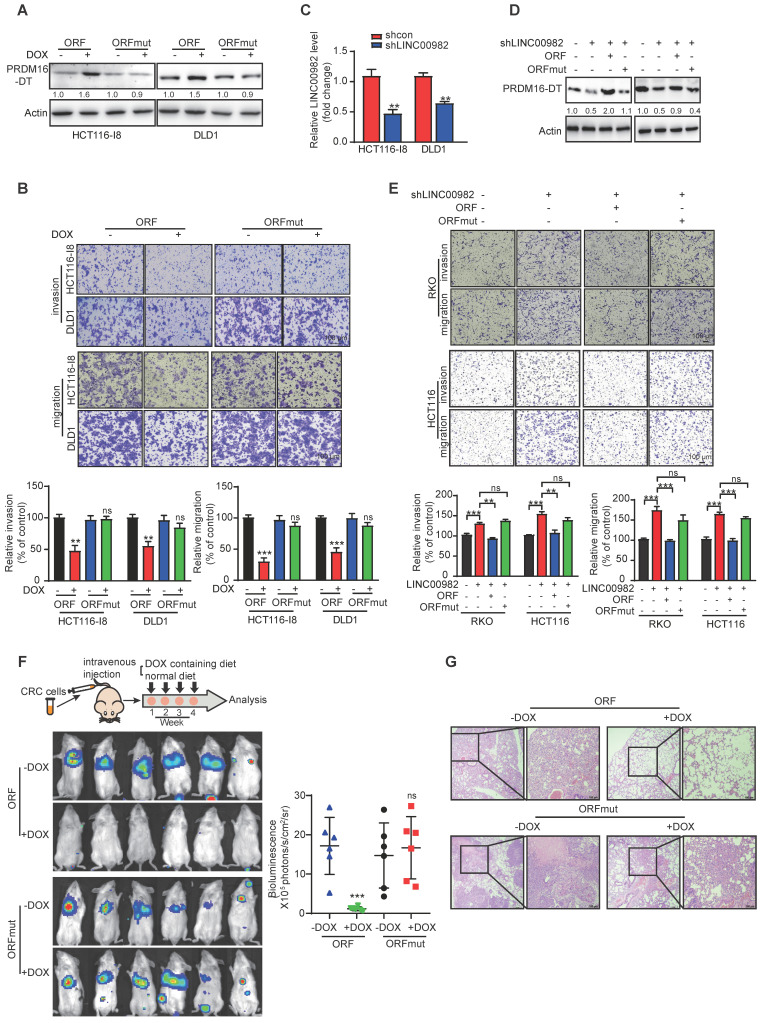
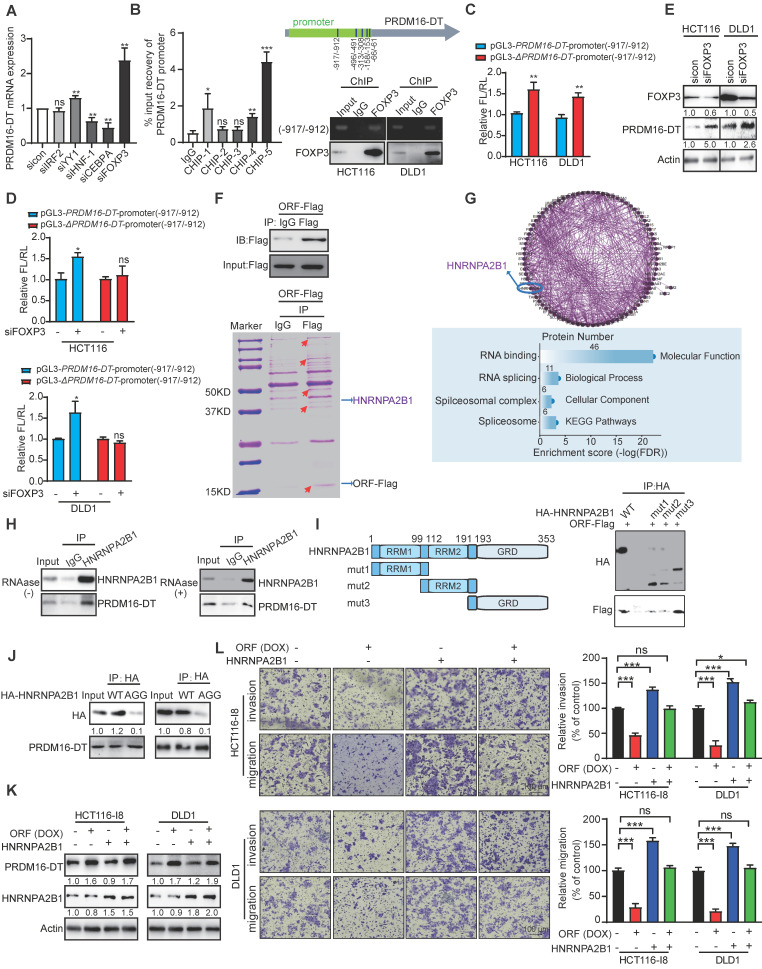
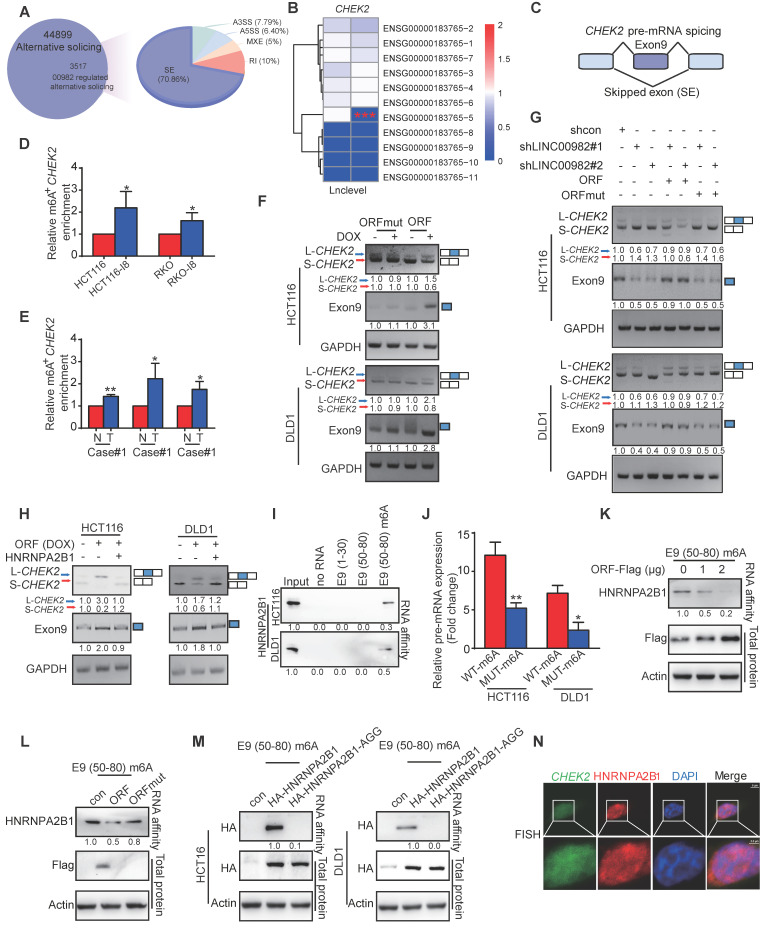
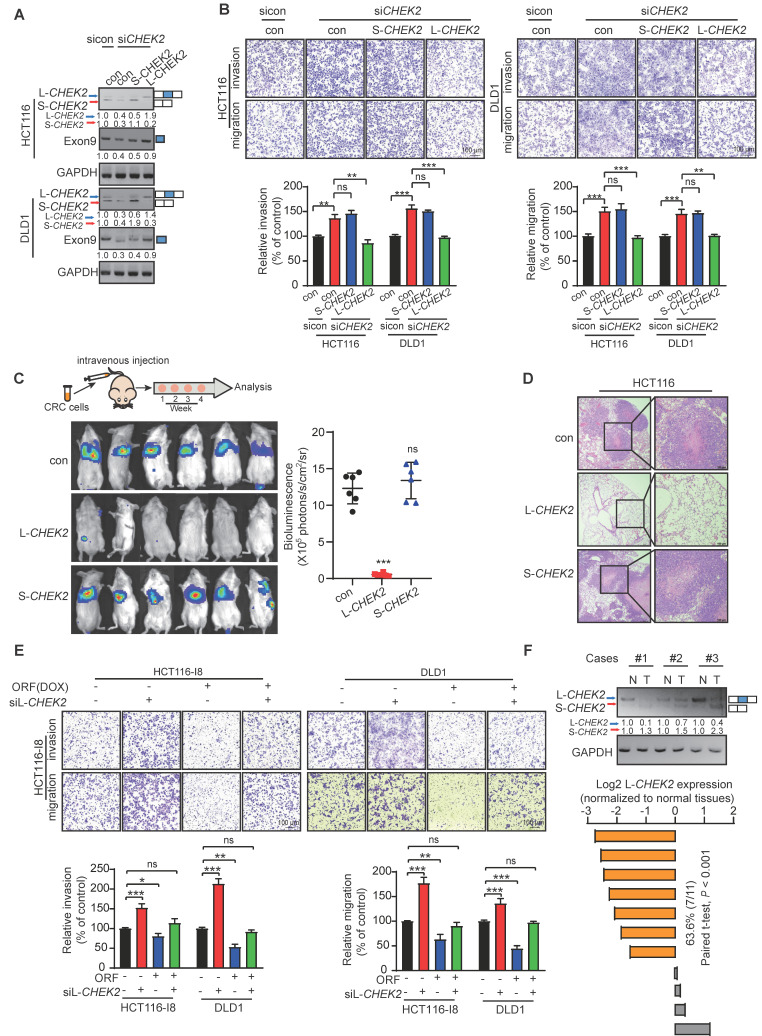
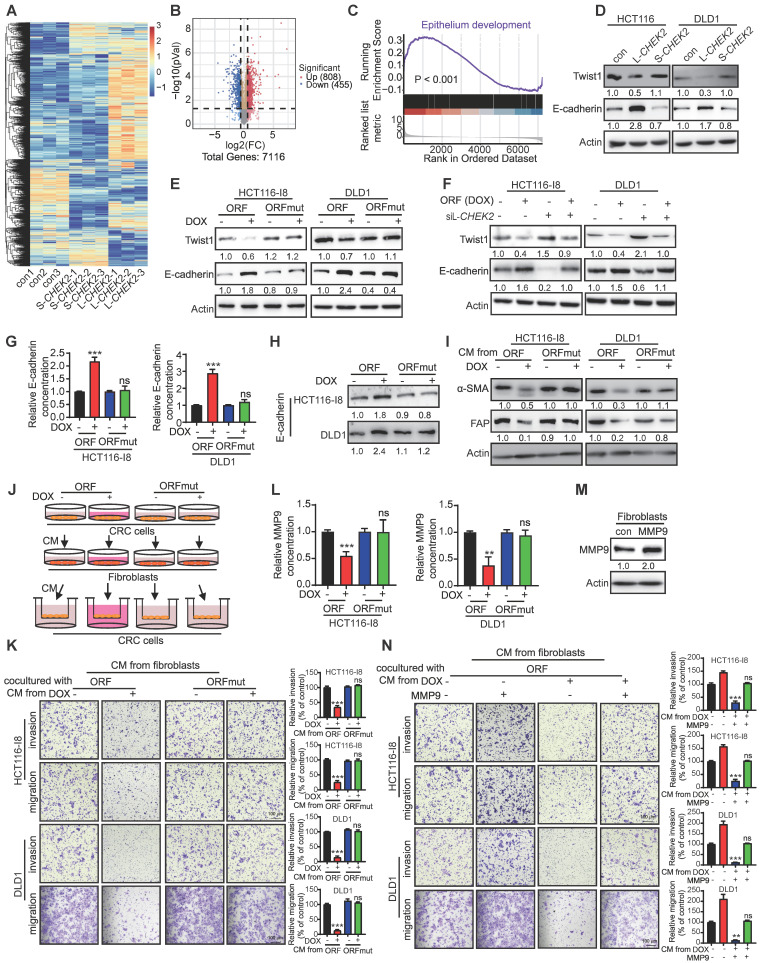
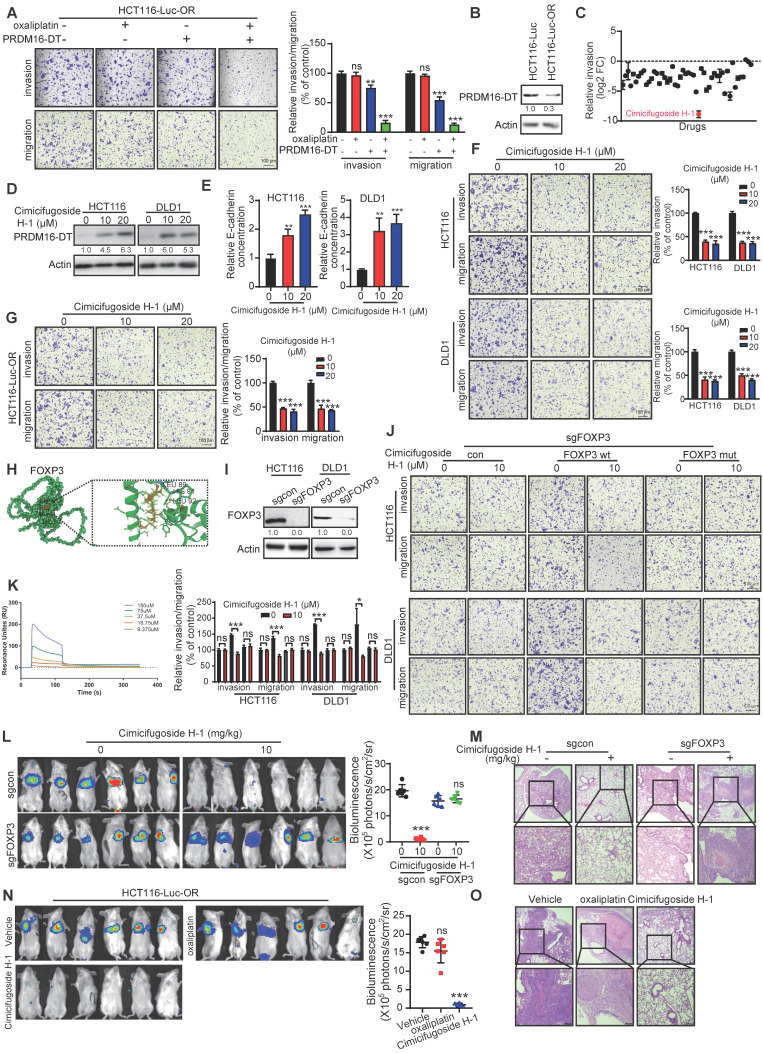
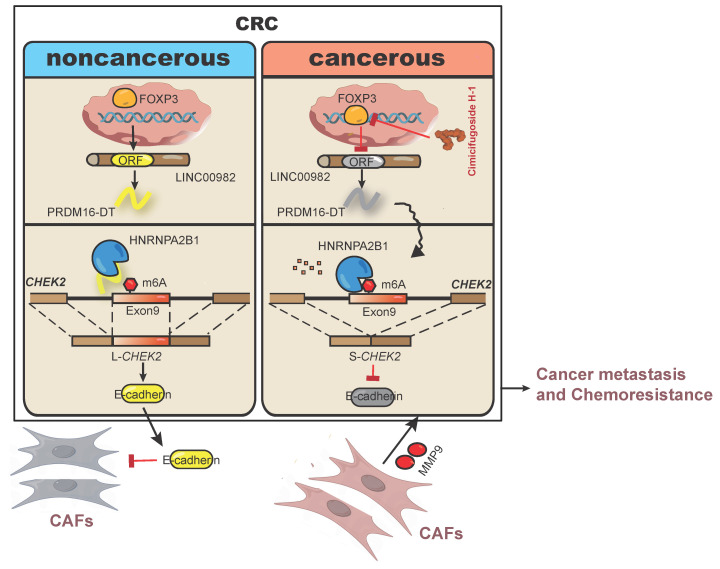
Metastasis is one of the key factors of treatment failure in late-stage colorectal cancer (CRC). Metastatic CRC frequently develops resistance to chemotherapeutic agents. This study aimed to identify the novel regulators from "hidden" proteins encoded by long noncoding RNAs (lncRNAs) involved in tumor metastasis and chemoresistance. Methods: CRISPR/Cas9 library functional screening was employed to identify the critical suppressor of cancer metastasis in highly invasive CRC models. Western blotting, immunofluorescence staining, invasion, migration, wound healing, WST-1, colony formation, gain- and loss-of-function experiments, in vivo experimental metastasis models, multiplex immunohistochemical staining, immunohistochemistry, qRT-PCR, and RT-PCR were used to assess the functional and clinical significance of FOXP3, PRDM16-DT, HNRNPA2B1, and L-CHEK2. RNA-sequencing, co-immunoprecipitation, qRT-PCR, RT-PCR, RNA affinity purification, RNA immunoprecipitation, MeRIP-quantitative PCR, fluorescence in situ hybridization, chromatin immunoprecipitation and luciferase reporter assay were performed to gain mechanistic insights into the role of PRDM16-DT in cancer metastasis and chemoresistance. An oxaliplatin-resistant CRC cell line was established by in vivo selection. WST-1, colony formation, invasion, migration, Biacore technology, gain- and loss-of-function experiments and an in vivo experimental metastasis model were used to determine the function and mechanism of cimicifugoside H-1 in CRC. Results: The novel protein PRDM16-DT, encoded by LINC00982, was identified as a cancer metastasis and chemoresistance suppressor. The down-regulated level of PRDM16-DT was positively associated with malignant phenotypes and poor prognosis of CRC patients. Transcriptionally regulated by FOXP3, PRDM16-DT directly interacted with HNRNPA2B1 and competitively decreased HNRNPA2B1 binding to exon 9 of CHEK2, resulting in the formation of long CHEK2 (L-CHEK2), subsequently promoting E-cadherin secretion. PRDM16-DT-induced E-cadherin secretion inhibited fibroblast activation, which in turn suppressed CRC metastasis by decreasing MMP9 secretion. Cimicifugoside H-1, a natural compound, can bind to LEU89, HIS91, and LEU92 of FOXP3 and significantly upregulated PRDM16-DT expression to repress CRC metastasis and reverse oxaliplatin resistance. Conclusions: lncRNA LINC00982 can express a new protein PRDM16-DT to function as a novel regulator in cancer metastasis and drug resistance of CRC. Cimicifugoside H-1 can act on the upstream of the PRDM16-DT signaling pathway to alleviate cancer chemoresistance.
Keywords: Cimicifugoside H-1; Colorectal cancer; Drug resistance.; Fibroblasts; PRDM16-DT.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Ciardiello F, Ciardiello D, Martini G, Napolitano S, Tabernero J, Cervantes A. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer J Clin. 2022;72(4):372–401. - PubMed
-
- Biller LH, Schrag D. Diagnosis and treatment of metastatic colorectal cancer: A Review. Jama. 2021;325(7):669–685. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
