

Pyruvate kinase M2 sustains cardiac mitochondrial quality surveillance in septic cardiomyopathy by regulating prohibitin 2 abundance via S91 phosphorylation
- PMID: 38856931
- PMCID: PMC11335292
- DOI: 10.1007/s00018-024-05253-9
Pyruvate kinase M2 sustains cardiac mitochondrial quality surveillance in septic cardiomyopathy by regulating prohibitin 2 abundance via S91 phosphorylation
Abstract
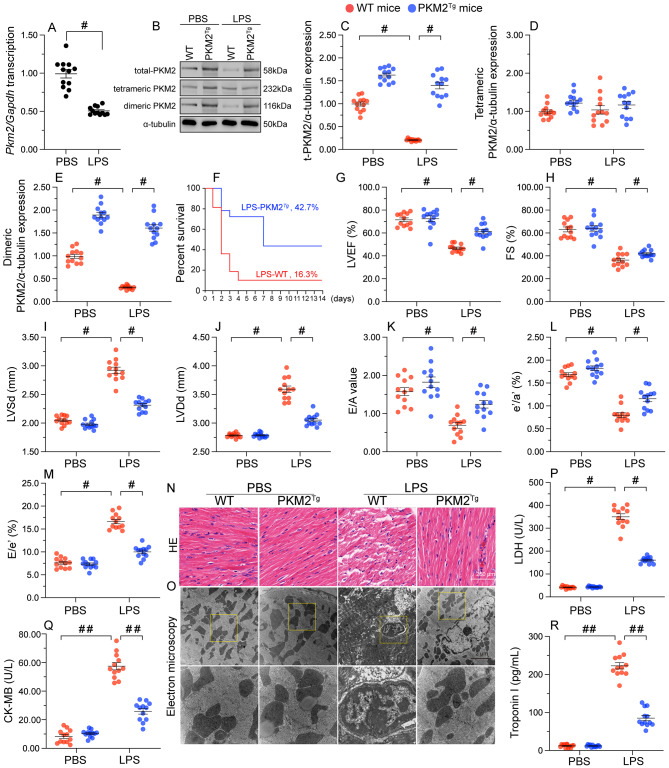
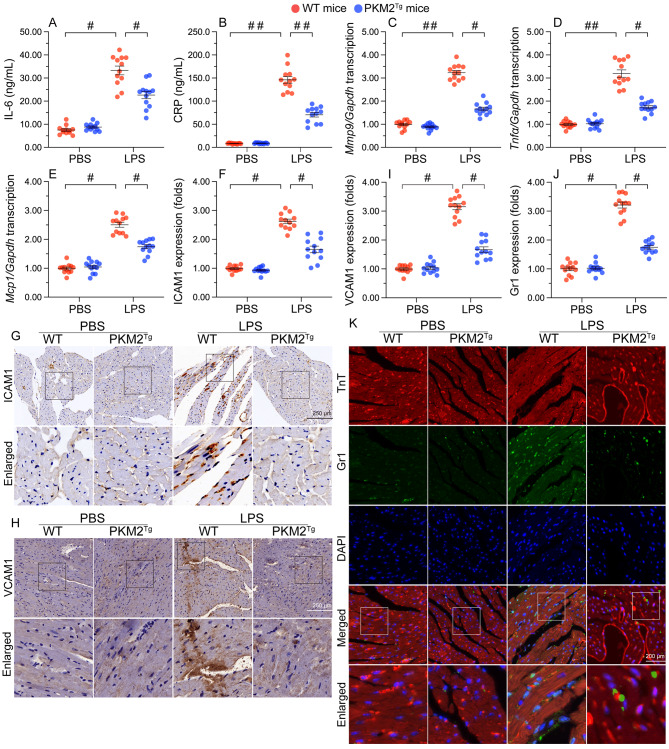
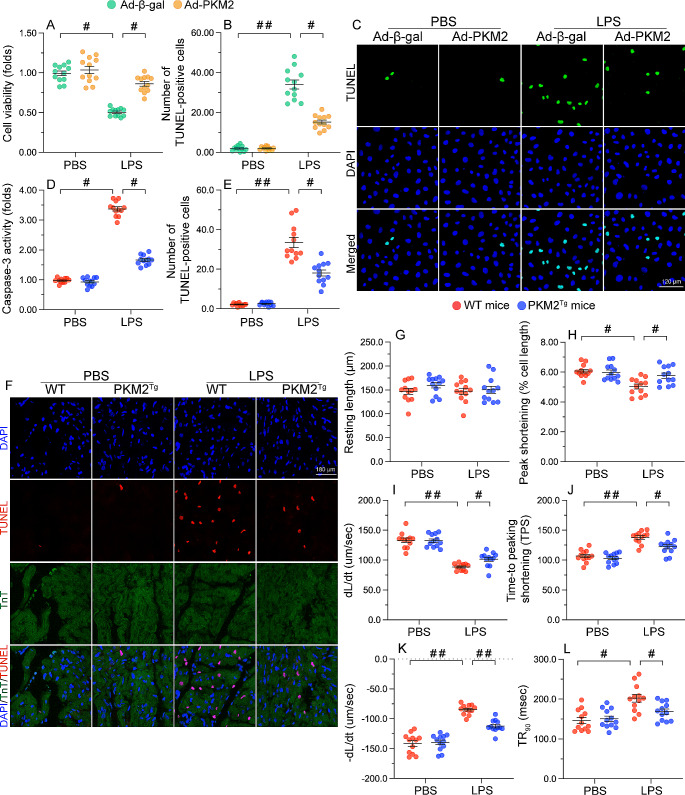
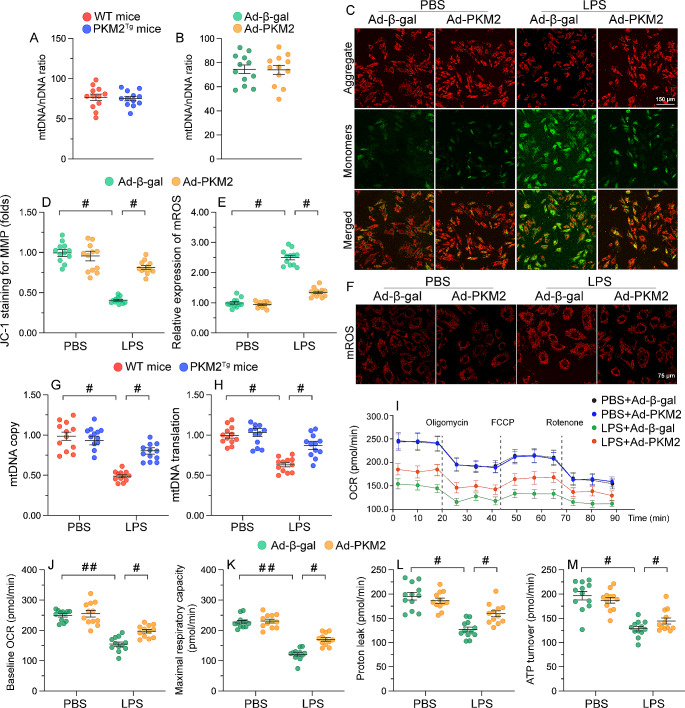
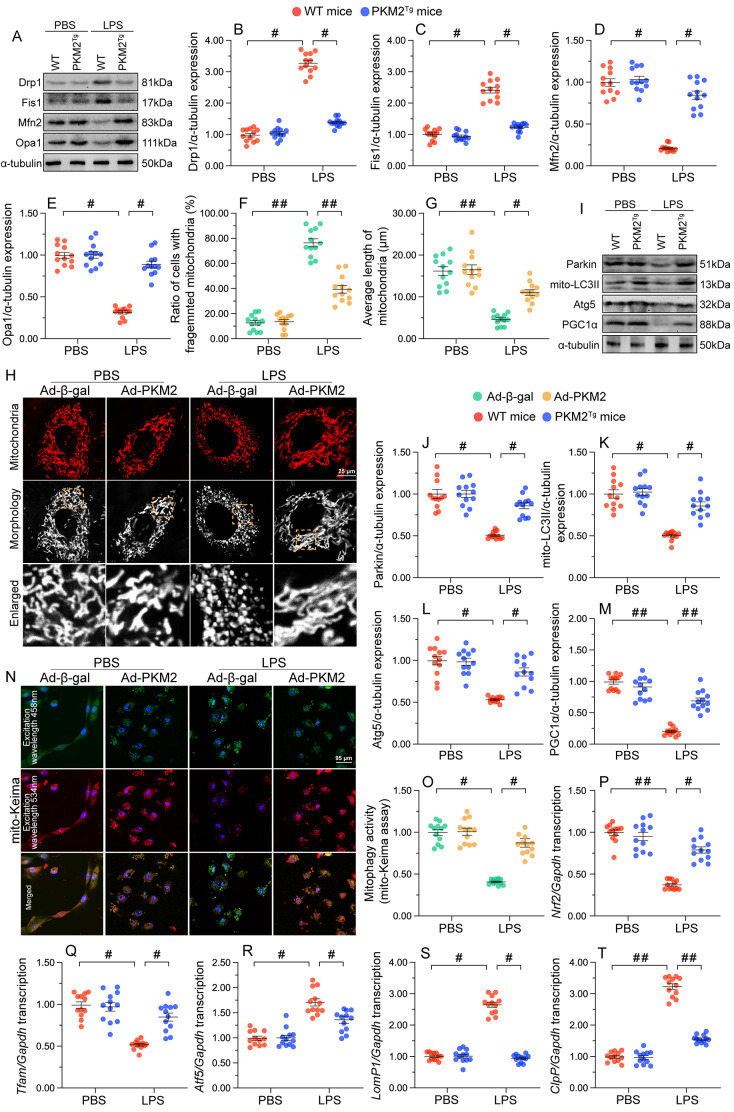
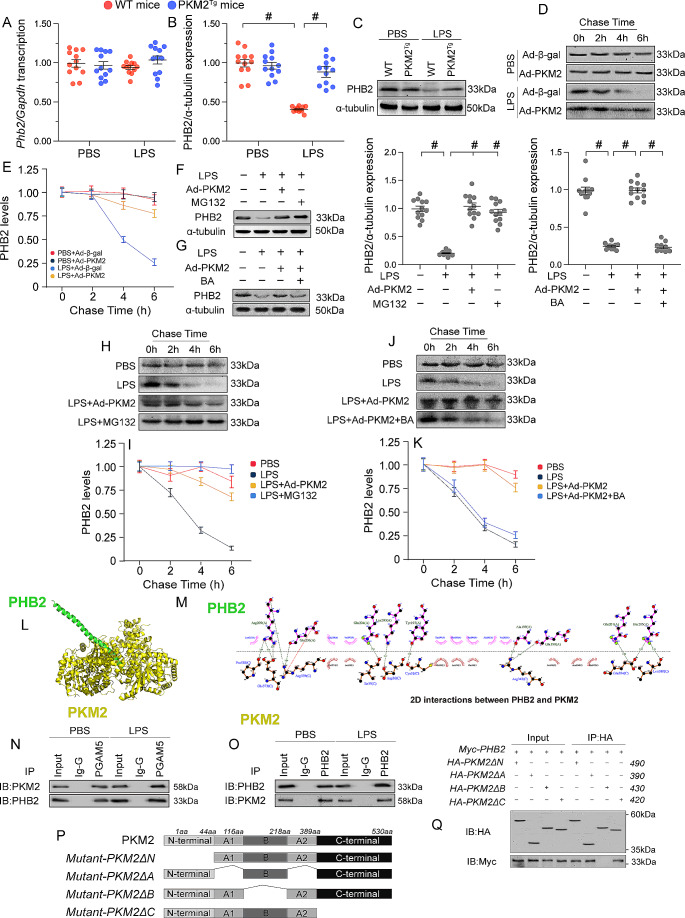
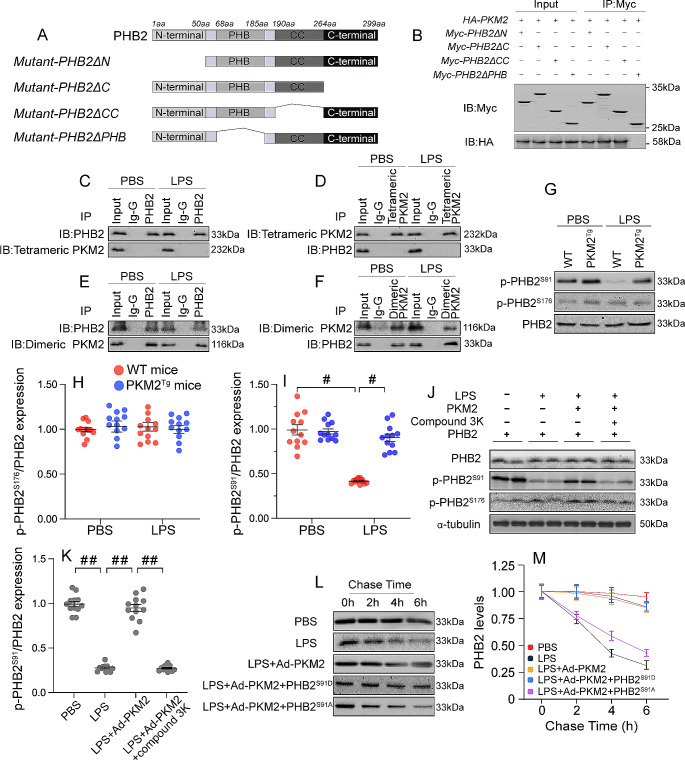
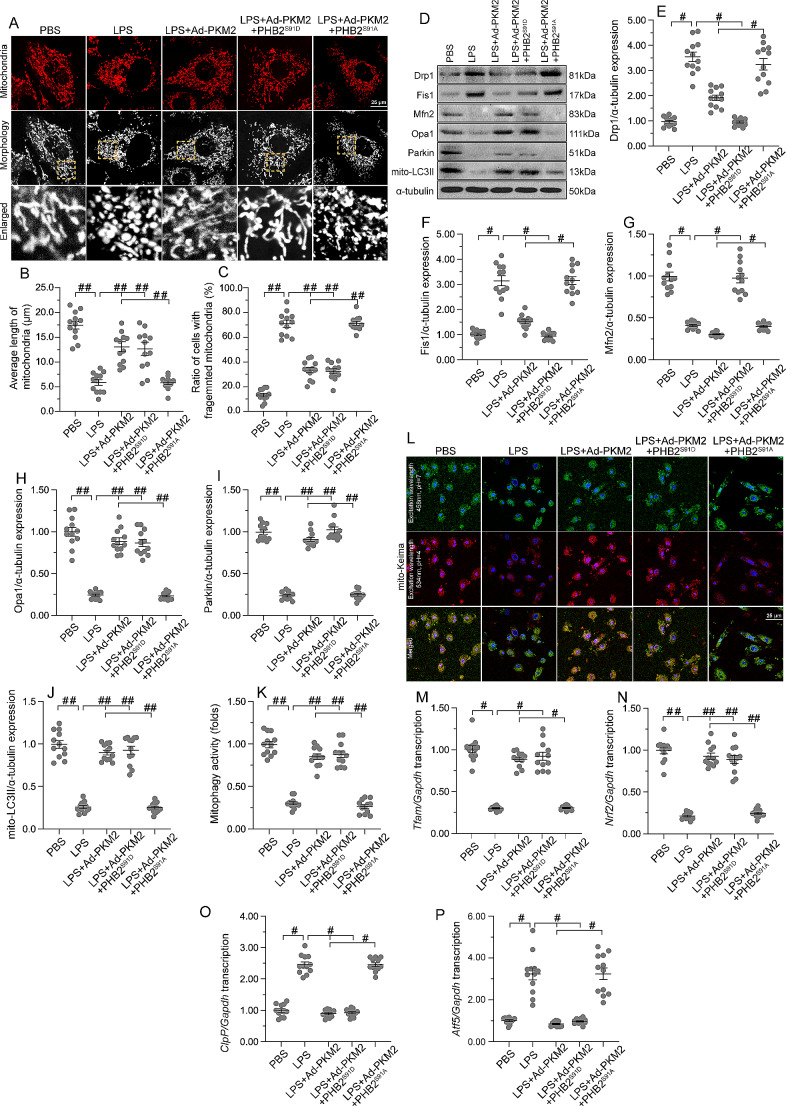
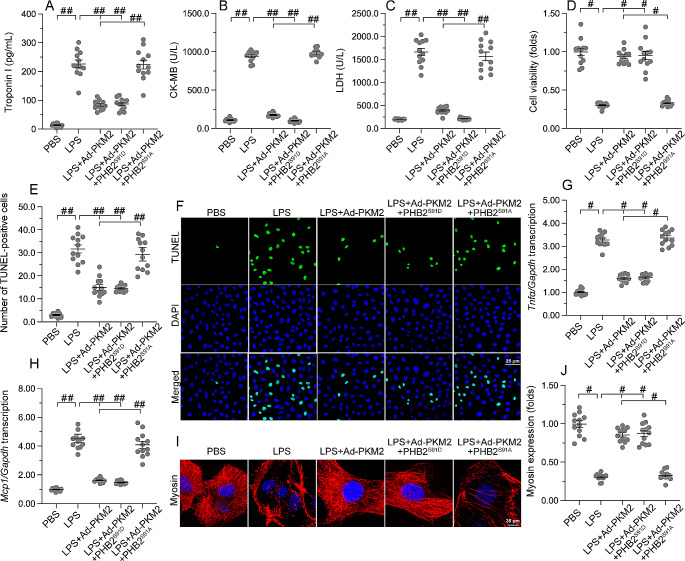
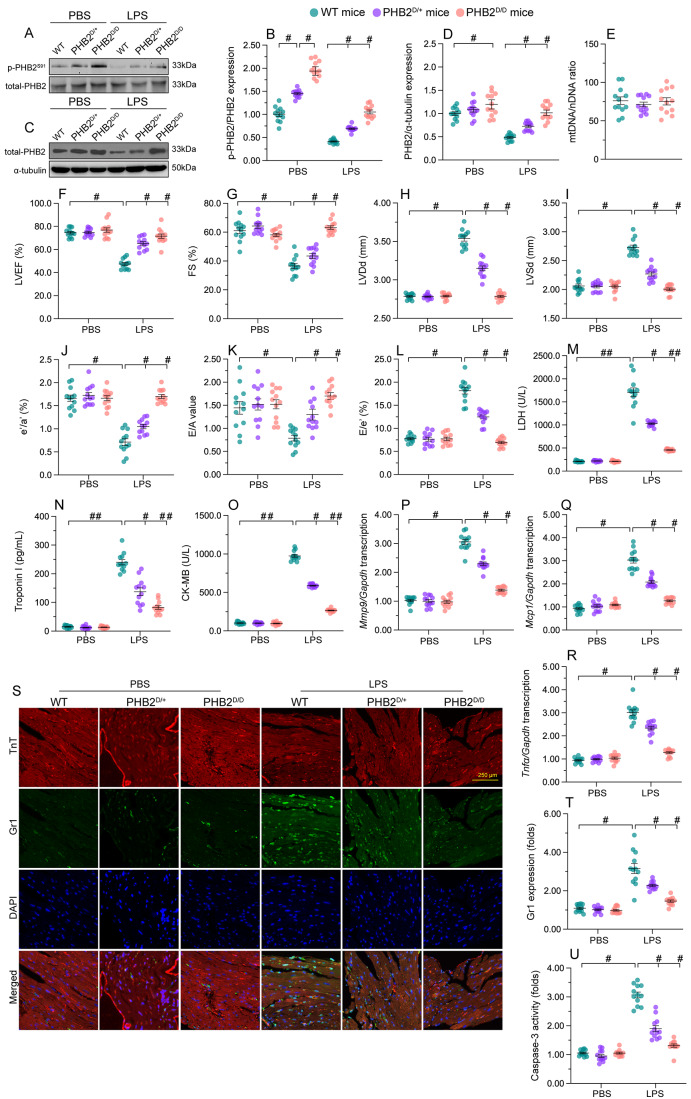
The endogenous mitochondrial quality control (MQC) system serves to protect mitochondria against cellular stressors. Although mitochondrial dysfunction contributes to cardiac damage during many pathological conditions, the regulatory signals influencing MQC disruption during septic cardiomyopathy (SC) remain unclear. This study aimed to investigate the involvement of pyruvate kinase M2 (PKM2) and prohibitin 2 (PHB2) interaction followed by MQC impairment in the pathogenesis of SC. We utilized LPS-induced SC models in PKM2 transgenic (PKM2TG) mice, PHB2S91D-knockin mice, and PKM2-overexpressing HL-1 cardiomyocytes. After LPS-induced SC, cardiac PKM2 expression was significantly downregulated in wild-type mice, whereas PKM2 overexpression in vivo sustained heart function, suppressed myocardial inflammation, and attenuated cardiomyocyte death. PKM2 overexpression relieved sepsis-related mitochondrial damage via MQC normalization, evidenced by balanced mitochondrial fission/fusion, activated mitophagy, restored mitochondrial biogenesis, and inhibited mitochondrial unfolded protein response. Docking simulations, co-IP, and domain deletion mutant protein transfection experiments showed that PKM2 phosphorylates PHB2 at Ser91, preventing LPS-mediated PHB2 degradation. Additionally, the A domain of PKM2 and the PHB domain of PHB2 are required for PKM2-PHB2 binding and PHB2 phosphorylation. After LPS exposure, expression of a phosphorylation-defective PHB2S91A mutant negated the protective effects of PKM2 overexpression. Moreover, knockin mice expressing a phosphorylation-mimetic PHB2S91D mutant showed improved heart function, reduced inflammation, and preserved mitochondrial function following sepsis induction. Abundant PKM2 expression is a prerequisite to sustain PKM2-PHB2 interaction which is a key element for preservation of PHB2 phosphorylation and MQC, presenting novel interventive targets for the treatment of septic cardiomyopathy.
Keywords: MQC; Mitochondria; PHB2; PKM2; Septic cardiomyopathy.
© 2024. The Author(s).
Conflict of interest statement
The authors declared no conflict of interest.
Figures
References
-
- Haileselassie B, Mukherjee R, Joshi AU, Napier BA, Massis LM, Ostberg NP, Queliconi BB, Monack D, Bernstein D, Mochly-Rosen D. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J Mol Cell Cardiol. 2019;130:160–169. doi: 10.1016/j.yjmcc.2019.04.006. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
