

Multi-cohort analysis reveals immune subtypes and predictive biomarkers in tuberculosis
- PMID: 38858405
- PMCID: PMC11164950
- DOI: 10.1038/s41598-024-63365-5
Multi-cohort analysis reveals immune subtypes and predictive biomarkers in tuberculosis
Abstract
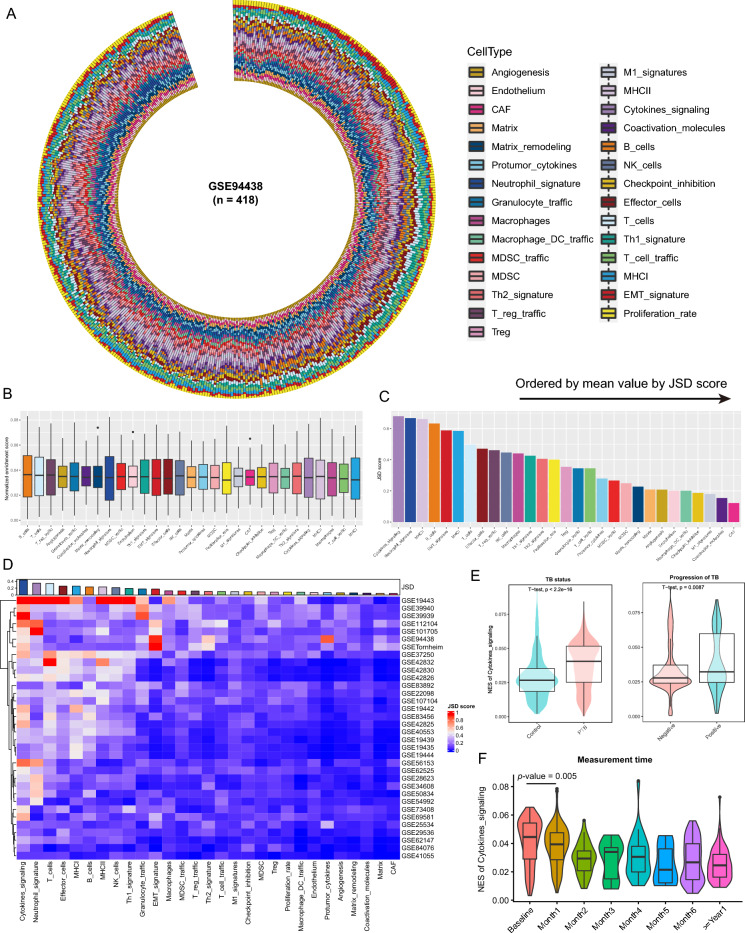
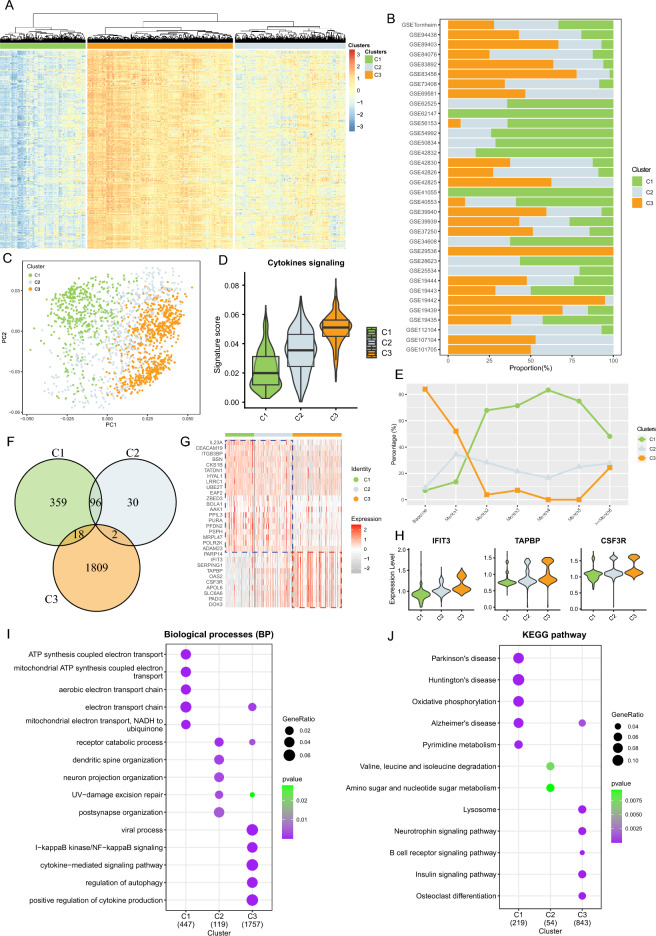
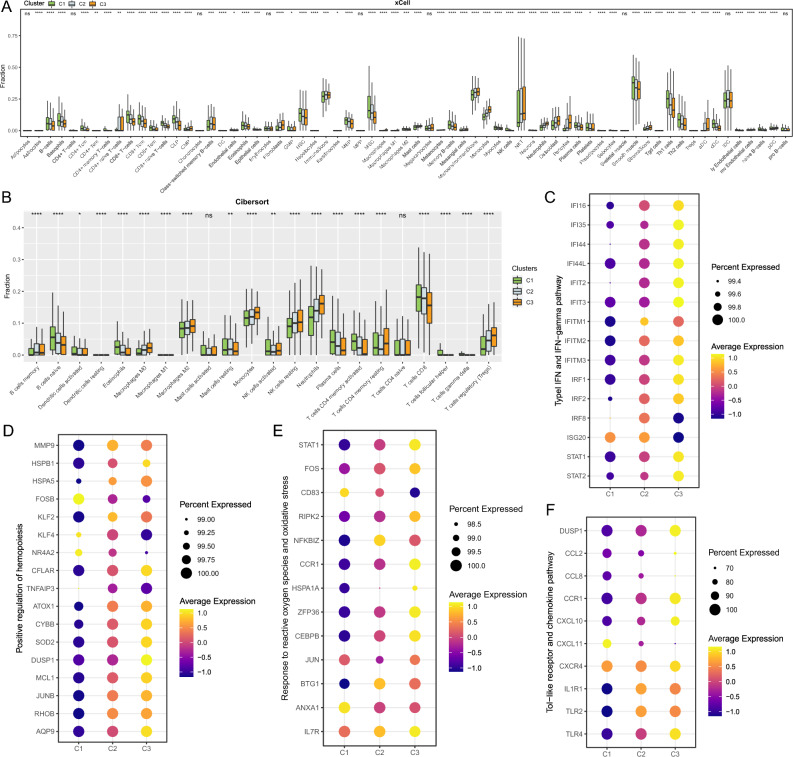
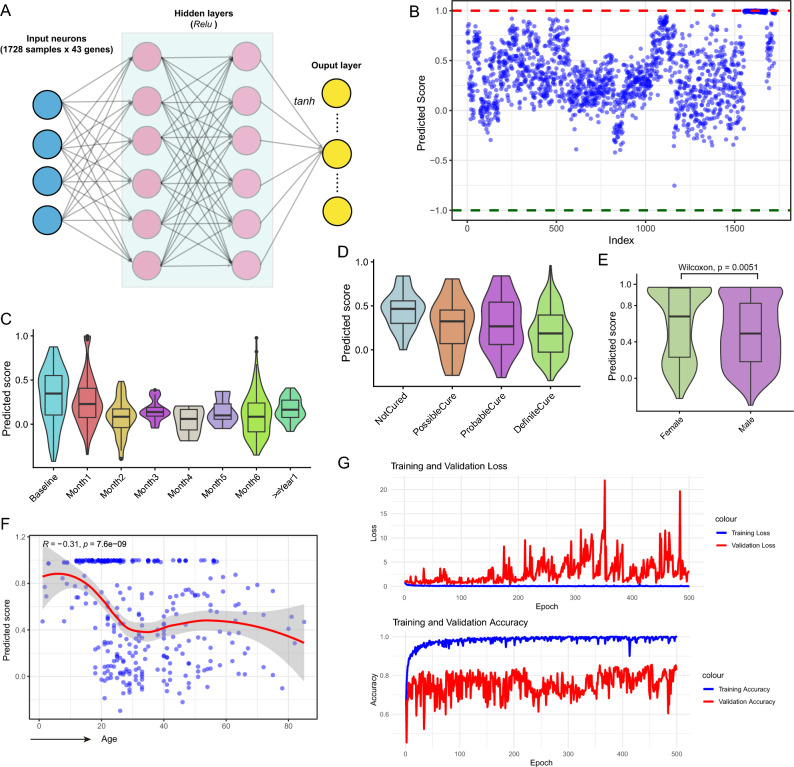
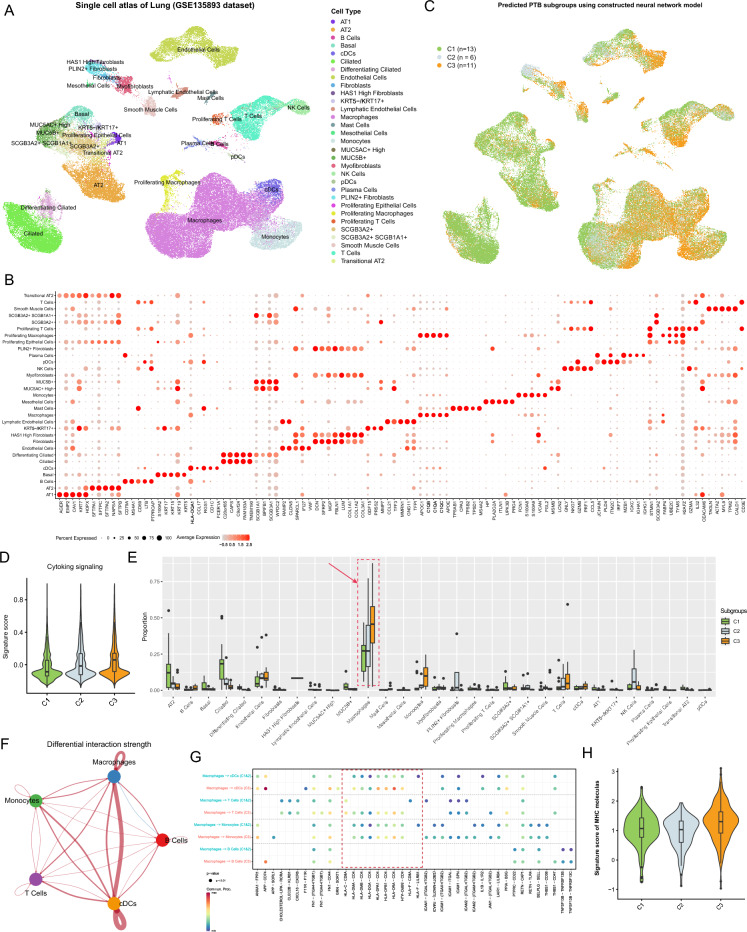
Tuberculosis (TB) remains a significant global health threat, necessitating effective strategies for diagnosis, prognosis, and treatment. This study employs a multi-cohort analysis approach to unravel the immune microenvironment of TB and delineate distinct subtypes within pulmonary TB (PTB) patients. Leveraging functional gene expression signatures (Fges), we identified three PTB subtypes (C1, C2, and C3) characterized by differential immune-inflammatory activity. These subtypes exhibited unique molecular features, functional disparities, and cell infiltration patterns, suggesting varying disease trajectories and treatment responses. A neural network model was developed to predict PTB progression based on a set of biomarker genes, achieving promising accuracy. Notably, despite both genders being affected by PTB, females exhibited a relatively higher risk of deterioration. Additionally, single-cell analysis provided insights into enhanced major histocompatibility complex (MHC) signaling in the rapid clearance of early pathogens in the C3 subgroup. This comprehensive approach offers valuable insights into PTB pathogenesis, facilitating personalized treatment strategies and precision medicine interventions.
Keywords: Immune microenvironment; Neural network model; PTB; Single-cell; Subtypes; Tuberculosis.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Mo, Y. et al. Bioinformatics analysis of new diagnostic and treatment biotargets in pulmonary tuberculosis.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
