

Systematic rare variant analyses identify RAB32 as a susceptibility gene for familial Parkinson's disease
- PMID: 38858457
- PMCID: PMC11250361
- DOI: 10.1038/s41588-024-01787-7
Systematic rare variant analyses identify RAB32 as a susceptibility gene for familial Parkinson's disease
Abstract
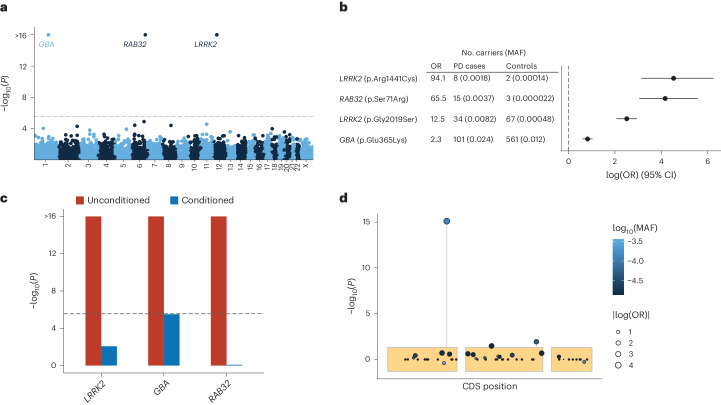
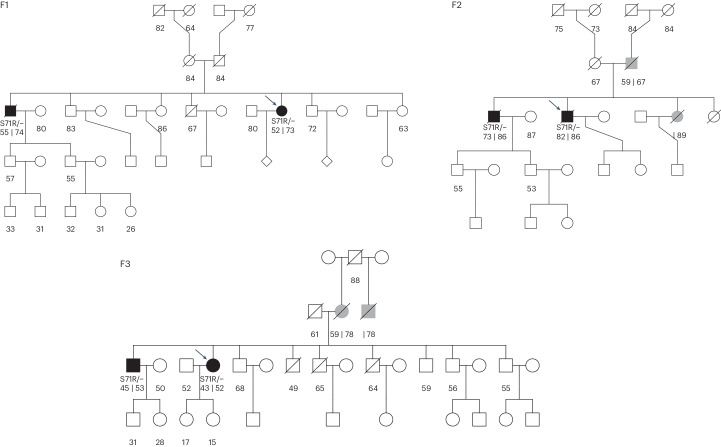
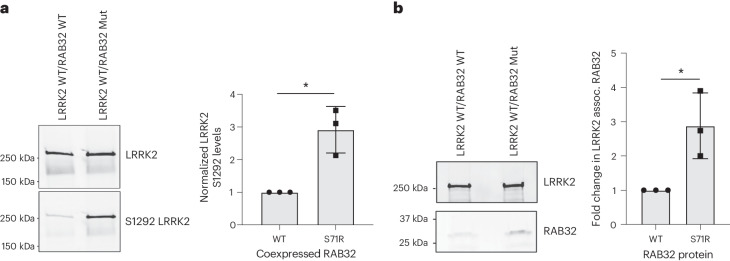
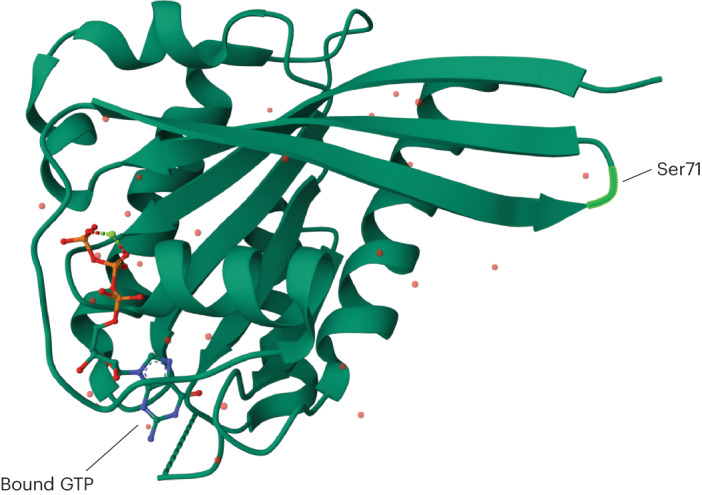
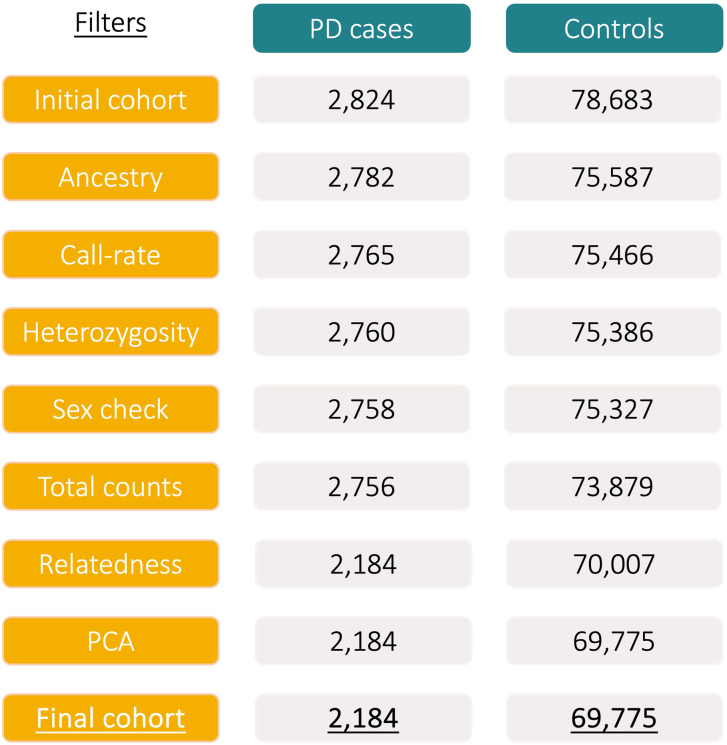
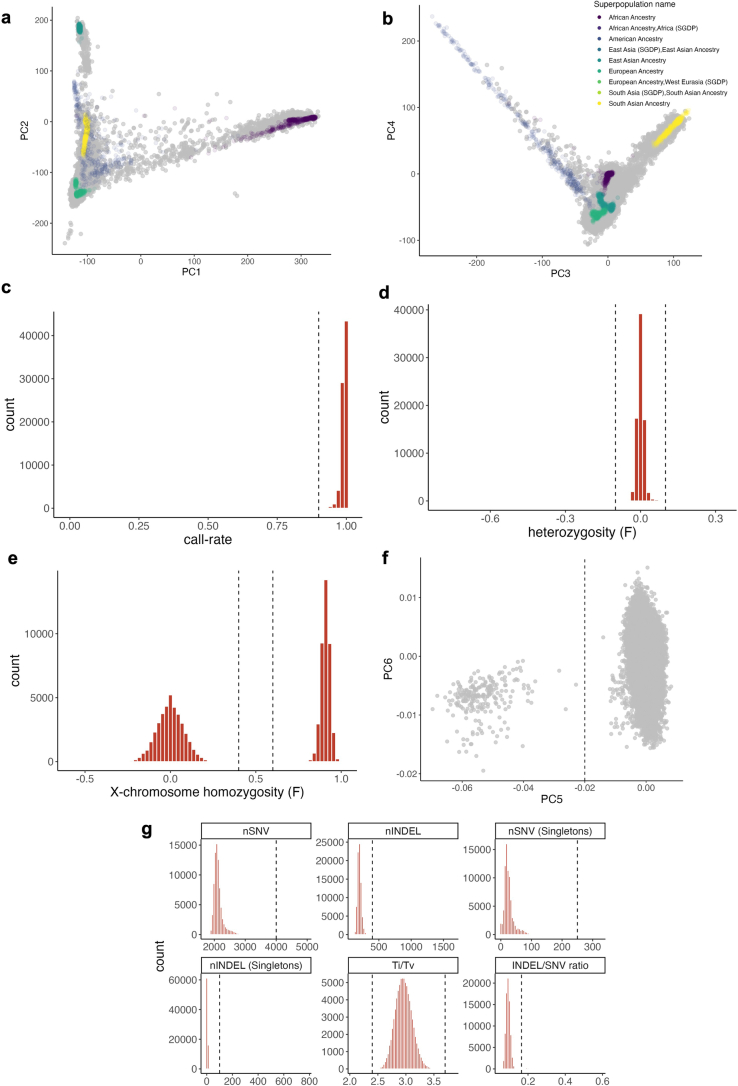
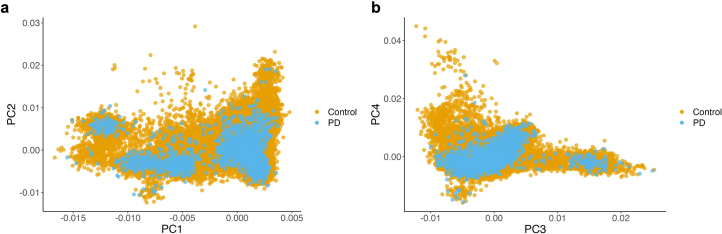
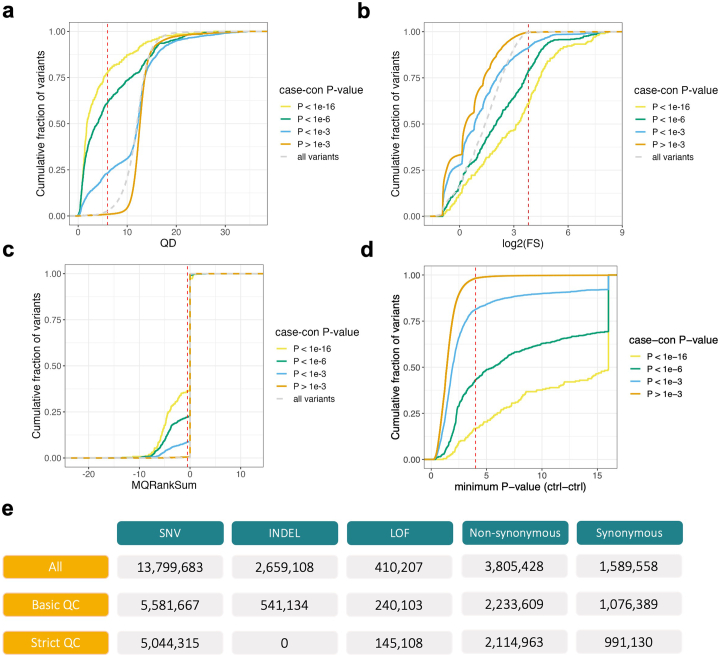
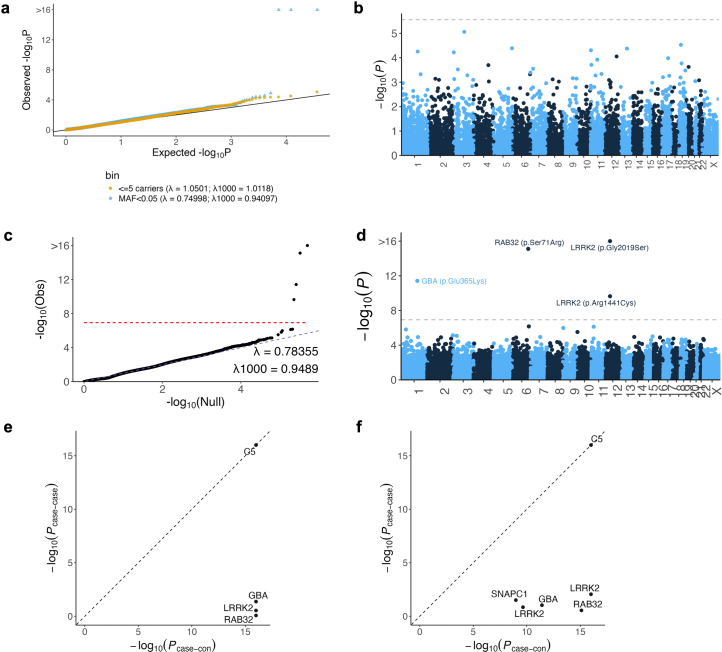
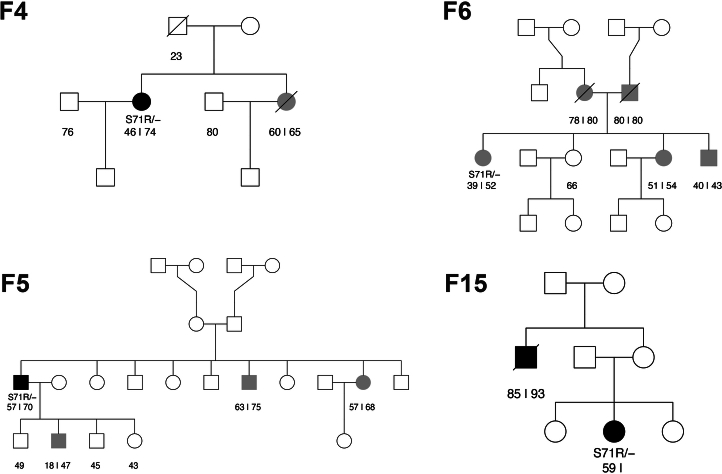
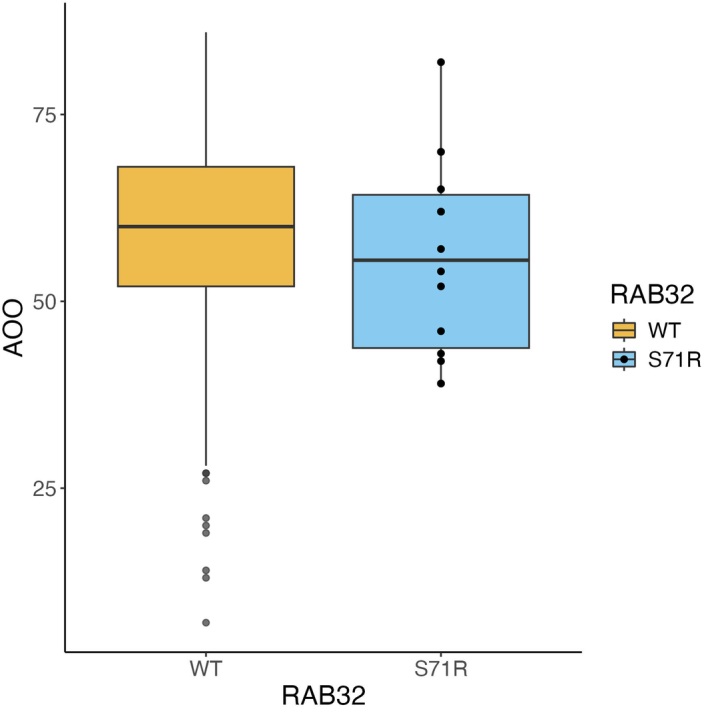
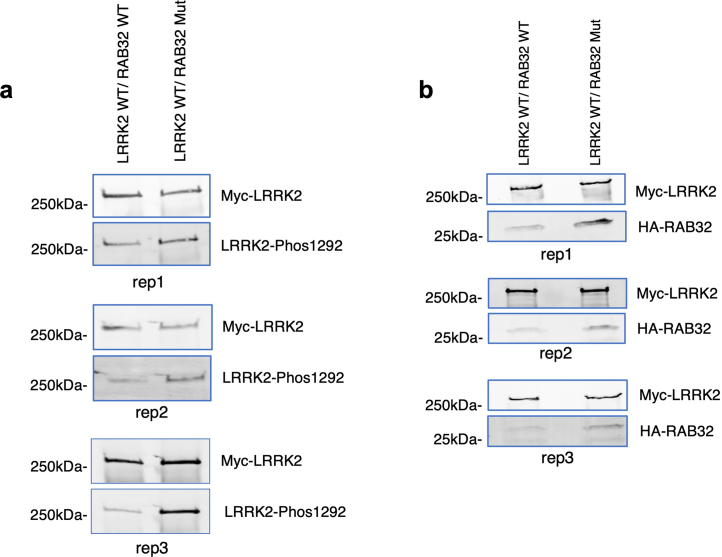
Despite substantial progress, causal variants are identified only for a minority of familial Parkinson's disease (PD) cases, leaving high-risk pathogenic variants unidentified1,2. To identify such variants, we uniformly processed exome sequencing data of 2,184 index familial PD cases and 69,775 controls. Exome-wide analyses converged on RAB32 as a novel PD gene identifying c.213C > G/p.S71R as a high-risk variant presenting in ~0.7% of familial PD cases while observed in only 0.004% of controls (odds ratio of 65.5). This variant was confirmed in all cases via Sanger sequencing and segregated with PD in three families. RAB32 encodes a small GTPase known to interact with LRRK2 (refs. 3,4). Functional analyses showed that RAB32 S71R increases LRRK2 kinase activity, as indicated by increased autophosphorylation of LRRK2 S1292. Here our results implicate mutant RAB32 in a key pathological mechanism in PD-LRRK2 kinase activity5-7-and thus provide novel insights into the mechanistic connections between RAB family biology, LRRK2 and PD risk.
© 2024. The Author(s).
Conflict of interest statement
Z.K.W. serves as principal investigator (PI) or co-PI on Biohaven Pharmaceuticals, Inc. (BHV4157-206) and Vigil Neuroscience, Inc. (VGL101-01.002, VGL101-01.201, positron emission tomography tracer development protocol, Csf1r biomarker and repository project, and ultrahigh field magnetic resonance imaging in the diagnosis and management of CSF1R-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia) projects/grants. He serves as co-PI of the Mayo Clinic APDA Center for Advanced Research and as an external advisory board member for the Vigil Neuroscience, Inc., and as a consultant on neurodegenerative medical research for Eli Lilli and Company. J.H.V. reports to have sponsored research agreements with Biogen and Astra Zeneca. J.E.L. is a consultant for Illios Therapeutics, WCG Clinical and Biogen. The other authors declare no competing interests.
Figures
Comment in
-
RAB32 variant is associated with PD susceptibility.Nat Rev Neurol. 2024 Aug;20(8):456. doi: 10.1038/s41582-024-00995-3. Nat Rev Neurol. 2024. PMID: 38977880 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
