

A viral E3 ubiquitin ligase produced by herpes simplex virus 1 inhibits the NLRP1 inflammasome
- PMID: 38861480
- PMCID: PMC11167375
- DOI: 10.1084/jem.20231518
A viral E3 ubiquitin ligase produced by herpes simplex virus 1 inhibits the NLRP1 inflammasome
Abstract
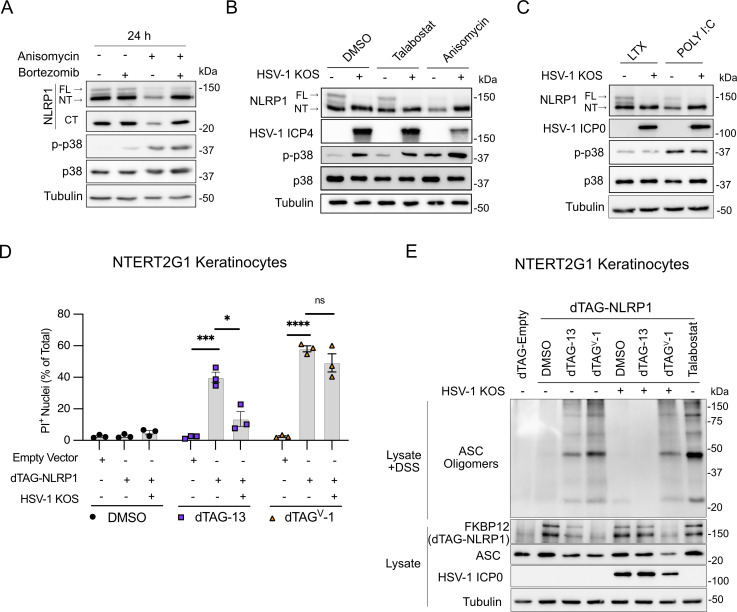
Guard proteins initiate defense mechanisms upon sensing pathogen-encoded virulence factors. Successful viral pathogens likely inhibit guard protein activity, but these interactions have been largely undefined. Here, we demonstrate that the human pathogen herpes simplex virus 1 (HSV-1) stimulates and inhibits an antiviral pathway initiated by NLRP1, a guard protein that induces inflammasome formation and pyroptotic cell death when activated. Notably, HSV-1 infection of human keratinocytes promotes posttranslational modifications to NLRP1, consistent with MAPK-dependent NLRP1 activation, but does not result in downstream inflammasome formation. We identify infected cell protein 0 (ICP0) as the critical HSV-1 protein that is necessary and sufficient for inhibition of the NLRP1 pathway. Mechanistically, ICP0's cytoplasmic localization and function as an E3 ubiquitin ligase prevents proteasomal degradation of the auto-inhibitory NT-NLRP1 fragment, thereby preventing inflammasome formation. Further, we demonstrate that inhibiting this inflammasome is important for promoting HSV-1 replication. Thus, we have established a mechanism by which HSV-1 overcomes a guard-mediated antiviral defense strategy in humans.
© 2024 Parameswaran et al.
Conflict of interest statement
Disclosures: The authors declare no competing interests exist.
Figures











Similar articles
-
Novel Role for Protein Inhibitor of Activated STAT 4 (PIAS4) in the Restriction of Herpes Simplex Virus 1 by the Cellular Intrinsic Antiviral Immune Response.J Virol. 2016 Apr 14;90(9):4807-4826. doi: 10.1128/JVI.03055-15. Print 2016 May. J Virol. 2016. PMID: 26937035 Free PMC article.
-
Characterization of Elements Regulating the Nuclear-to-Cytoplasmic Translocation of ICP0 in Late Herpes Simplex Virus 1 Infection.J Virol. 2018 Jan 2;92(2):e01673-17. doi: 10.1128/JVI.01673-17. Print 2018 Jan 15. J Virol. 2018. PMID: 29093084 Free PMC article.
-
Discovery of Small-Molecule Inhibitors Targeting the E3 Ubiquitin Ligase Activity of the Herpes Simplex Virus 1 ICP0 Protein Using an In Vitro High-Throughput Screening Assay.J Virol. 2019 Jun 14;93(13):e00619-19. doi: 10.1128/JVI.00619-19. Print 2019 Jul 1. J Virol. 2019. PMID: 30996104 Free PMC article.
-
The NLRP1 Inflammasome in Human Skin and Beyond.Int J Mol Sci. 2020 Jul 6;21(13):4788. doi: 10.3390/ijms21134788. Int J Mol Sci. 2020. PMID: 32640751 Free PMC article. Review.
-
Tripping the wire: sensing of viral protease activity by CARD8 and NLRP1 inflammasomes.Curr Opin Immunol. 2023 Aug;83:102354. doi: 10.1016/j.coi.2023.102354. Epub 2023 Jun 11. Curr Opin Immunol. 2023. PMID: 37311351 Free PMC article. Review.
Cited by
-
The precise function of alphaherpesvirus tegument proteins and their interactions during the viral life cycle.Front Microbiol. 2024 Jul 2;15:1431672. doi: 10.3389/fmicb.2024.1431672. eCollection 2024. Front Microbiol. 2024. PMID: 39015737 Free PMC article. Review.
-
Next-generation replication-defective HSV vectors for delivery of large DNA payloads.Mol Ther. 2025 May 7;33(5):2205-2216. doi: 10.1016/j.ymthe.2025.03.055. Epub 2025 Apr 2. Mol Ther. 2025. PMID: 40181547 Review.
-
Portimine A toxin causes skin inflammation through ZAKα-dependent NLRP1 inflammasome activation.EMBO Mol Med. 2025 Mar;17(3):535-562. doi: 10.1038/s44321-025-00197-4. Epub 2025 Feb 13. EMBO Mol Med. 2025. PMID: 39948420 Free PMC article.
-
Ubiquitination Regulates Reorganization of the Membrane System During Cytomegalovirus Infection.Life (Basel). 2025 Jul 31;15(8):1212. doi: 10.3390/life15081212. Life (Basel). 2025. PMID: 40868860 Free PMC article.
References
-
- Ball, D.P., Taabazuing C.Y., Griswold A.R., Orth E.L., Rao S.D., Kotliar I.B., Johnson D.C., and Bachovchin D.A.. 2019. Human caspase-1 autoproteolysis is required for ASC-dependent and -independent inflammasome activation. bioRxiv. 10.1101/681304 (Preprint posted June 24, 2019). - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
