

Modulation of FGF pathway signaling and vascular differentiation using designed oligomeric assemblies
- PMID: 38861993
- PMCID: PMC11246234
- DOI: 10.1016/j.cell.2024.05.025
Modulation of FGF pathway signaling and vascular differentiation using designed oligomeric assemblies
Abstract
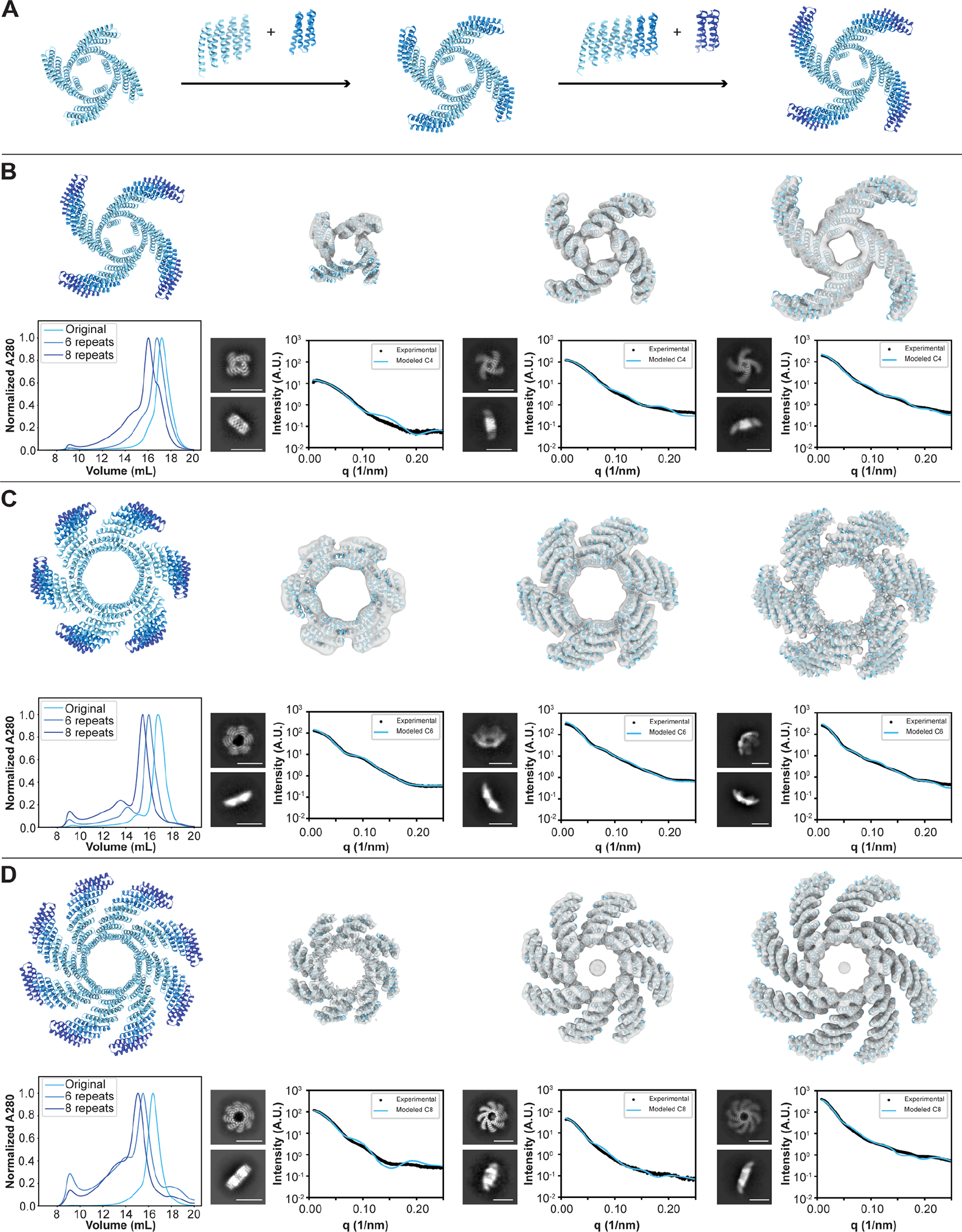
Many growth factors and cytokines signal by binding to the extracellular domains of their receptors and driving association and transphosphorylation of the receptor intracellular tyrosine kinase domains, initiating downstream signaling cascades. To enable systematic exploration of how receptor valency and geometry affect signaling outcomes, we designed cyclic homo-oligomers with up to 8 subunits using repeat protein building blocks that can be modularly extended. By incorporating a de novo-designed fibroblast growth factor receptor (FGFR)-binding module into these scaffolds, we generated a series of synthetic signaling ligands that exhibit potent valency- and geometry-dependent Ca2+ release and mitogen-activated protein kinase (MAPK) pathway activation. The high specificity of the designed agonists reveals distinct roles for two FGFR splice variants in driving arterial endothelium and perivascular cell fates during early vascular development. Our designed modular assemblies should be broadly useful for unraveling the complexities of signaling in key developmental transitions and for developing future therapeutic applications.
Keywords: FGF signaling; binder design; cryo-EM; de novo protein design; endothelial cell differentiation; iPSCs; oligomeric scaffolds; vascular development.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests L.S., D.B., N.I.E., T.S., A.E., A.F., W.Y., D.R.H., B.C., H.R.-B., A.P., R.L.R., G.B., and D.C.E. are coinventors on a patent application that has been filed by the University of Washington.
Figures






Update of
-
Modulation of FGF pathway signaling and vascular differentiation using designed oligomeric assemblies.bioRxiv [Preprint]. 2023 Mar 15:2023.03.14.532666. doi: 10.1101/2023.03.14.532666. bioRxiv. 2023. Update in: Cell. 2024 Jul 11;187(14):3726-3740.e43. doi: 10.1016/j.cell.2024.05.025. PMID: 36993355 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
- R01 DE033016/DE/NIDCR NIH HHS/United States
- P30 GM124169/GM/NIGMS NIH HHS/United States
- R01 AG063845/AG/NIA NIH HHS/United States
- S10 OD018483/OD/NIH HHS/United States
- R35 GM150919/GM/NIGMS NIH HHS/United States
- U01 HL099993/HL/NHLBI NIH HHS/United States
- U01 HL099997/HL/NHLBI NIH HHS/United States
- U19 AG065156/AG/NIA NIH HHS/United States
- R01 GM097372/GM/NIGMS NIH HHS/United States
- R35 GM128777/GM/NIGMS NIH HHS/United States
- T90 DE021984/DE/NIDCR NIH HHS/United States
- R01 GM083867/GM/NIGMS NIH HHS/United States
- P01 GM081619/GM/NIGMS NIH HHS/United States
- U24 GM129539/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
