

Regulated cell death in hypoxic-ischaemic encephalopathy: recent development and mechanistic overview
- PMID: 38862503
- PMCID: PMC11167026
- DOI: 10.1038/s41420-024-02014-2
Regulated cell death in hypoxic-ischaemic encephalopathy: recent development and mechanistic overview
Abstract
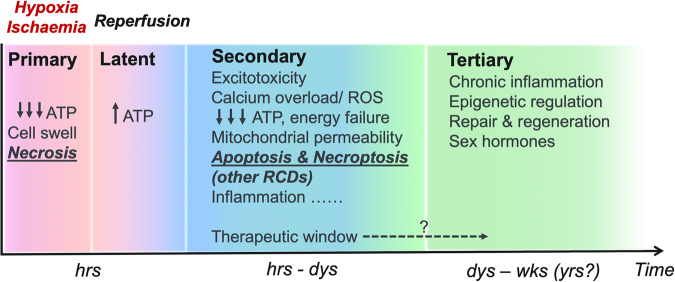
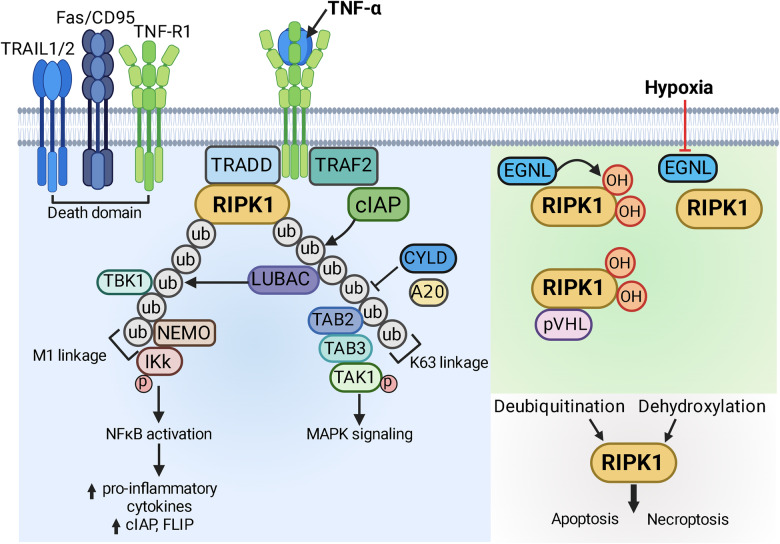
Hypoxic-ischaemic encephalopathy (HIE) in termed infants remains a significant cause of morbidity and mortality worldwide despite the introduction of therapeutic hypothermia. Depending on the cell type, cellular context, metabolic predisposition and insult severity, cell death in the injured immature brain can be highly heterogenous. A continuum of cell death exists in the H/I-injured immature brain. Aside from apoptosis, emerging evidence supports the pathological activation of necroptosis, pyroptosis and ferroptosis as alternative regulated cell death (RCD) in HIE to trigger neuroinflammation and metabolic disturbances in addition to cell loss. Upregulation of autophagy and mitophagy in HIE represents an intrinsic neuroprotective strategy. Molecular crosstalk between RCD pathways implies one RCD mechanism may compensate for the loss of function of another. Moreover, mitochondrion was identified as the signalling "hub" where different RCD pathways converge. The highly-orchestrated nature of RCD makes them promising therapeutic targets. Better understanding of RCD mechanisms and crosstalk between RCD subtypes likely shed light on novel therapy development for HIE. The identification of a potential RCD converging node may open up the opportunity for simultaneous and synergistic inhibition of cell death in the immature brain.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures






Similar articles
-
Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research.J Hematol Oncol. 2022 Dec 8;15(1):174. doi: 10.1186/s13045-022-01392-3. J Hematol Oncol. 2022. PMID: 36482419 Free PMC article. Review.
-
Emerging role of necroptosis, pyroptosis, and ferroptosis in breast cancer: New dawn for overcoming therapy resistance.Neoplasia. 2024 Sep;55:101017. doi: 10.1016/j.neo.2024.101017. Epub 2024 Jun 14. Neoplasia. 2024. PMID: 38878618 Free PMC article. Review.
-
Targeting novel regulated cell death:Ferroptosis, pyroptosis, and autophagy in sepsis-associated encephalopathy.Biomed Pharmacother. 2024 May;174:116453. doi: 10.1016/j.biopha.2024.116453. Epub 2024 Mar 20. Biomed Pharmacother. 2024. PMID: 38513593 Review.
-
Targeting regulated cell death (RCD) with small-molecule compounds in triple-negative breast cancer: a revisited perspective from molecular mechanisms to targeted therapies.J Hematol Oncol. 2022 Apr 12;15(1):44. doi: 10.1186/s13045-022-01260-0. J Hematol Oncol. 2022. PMID: 35414025 Free PMC article. Review.
-
Regulated cell death: discovery, features and implications for neurodegenerative diseases.Cell Commun Signal. 2021 Dec 18;19(1):120. doi: 10.1186/s12964-021-00799-8. Cell Commun Signal. 2021. PMID: 34922574 Free PMC article. Review.
Cited by
-
Hypothermia regulates mitophagy and apoptosis via PINK1/Parkin-VDAC 3 signaling pathway during oxygen-glucose deprivation/recovery injury.Sci Rep. 2025 Feb 7;15(1):4607. doi: 10.1038/s41598-025-89176-w. Sci Rep. 2025. PMID: 39920327 Free PMC article.
-
RORα-activated mitophagy attenuating hypoxic-ischemic encephalopathy via suppression of microglial cGAS-STING axis.Front Immunol. 2025 Jul 29;16:1592737. doi: 10.3389/fimmu.2025.1592737. eCollection 2025. Front Immunol. 2025. PMID: 40799648 Free PMC article.
-
miRNA-105 Attenuates Hypoxic-Ischemic Brain Damage in Neonatal Rats by Inhibiting Apoptosis and Necroptosis.Neurochem Res. 2025 Jul 21;50(4):241. doi: 10.1007/s11064-025-04484-x. Neurochem Res. 2025. PMID: 40690061
-
Diagnosis of Multiple Organ Dysfunction in Neonates with Hypoxic-Ischemic Encephalopathy: Vasoactive Inotropic Score, Renal Score, Fibrosis-5 Index and Lactate/Albumin Ratio.Diagnostics (Basel). 2024 Dec 12;14(24):2796. doi: 10.3390/diagnostics14242796. Diagnostics (Basel). 2024. PMID: 39767157 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
