

Mechanistic analysis of enhancer sequences in the estrogen receptor transcriptional program
- PMID: 38862711
- PMCID: PMC11167054
- DOI: 10.1038/s42003-024-06400-5
Mechanistic analysis of enhancer sequences in the estrogen receptor transcriptional program
Abstract
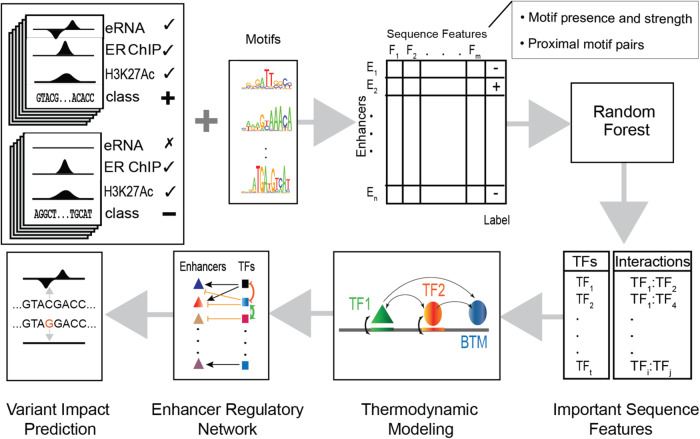
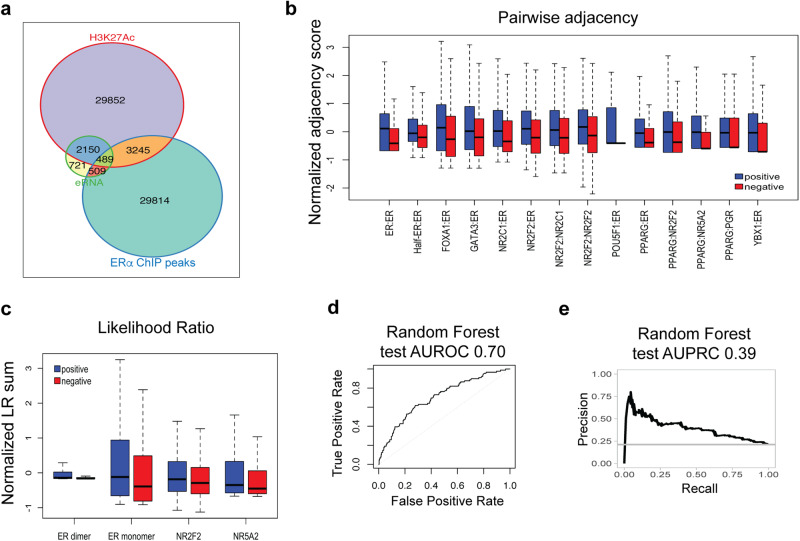
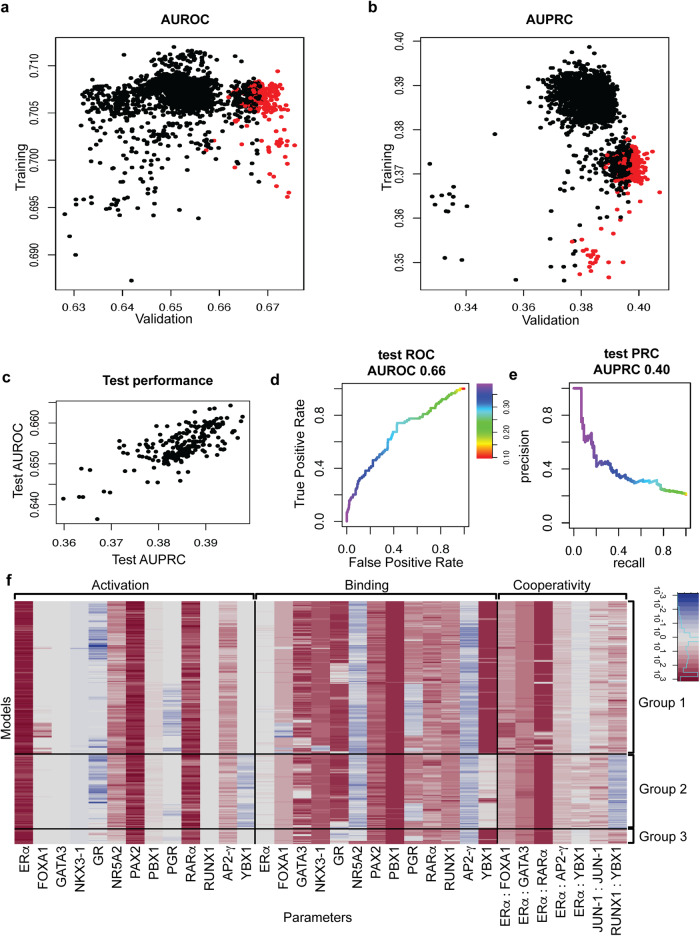
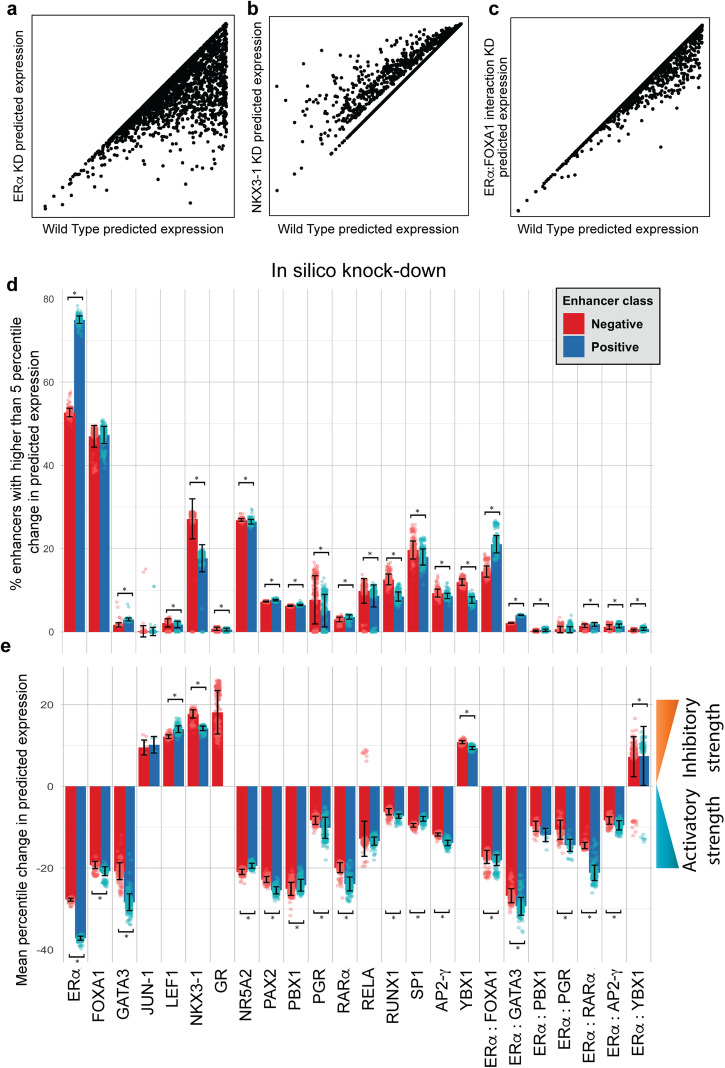
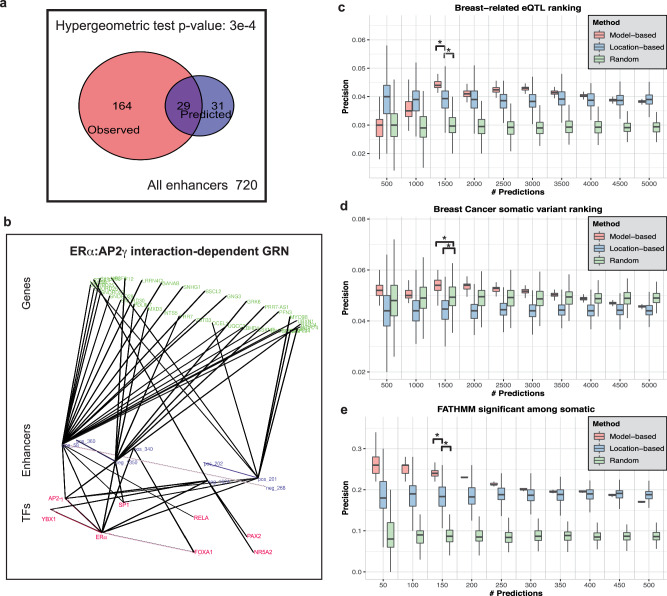
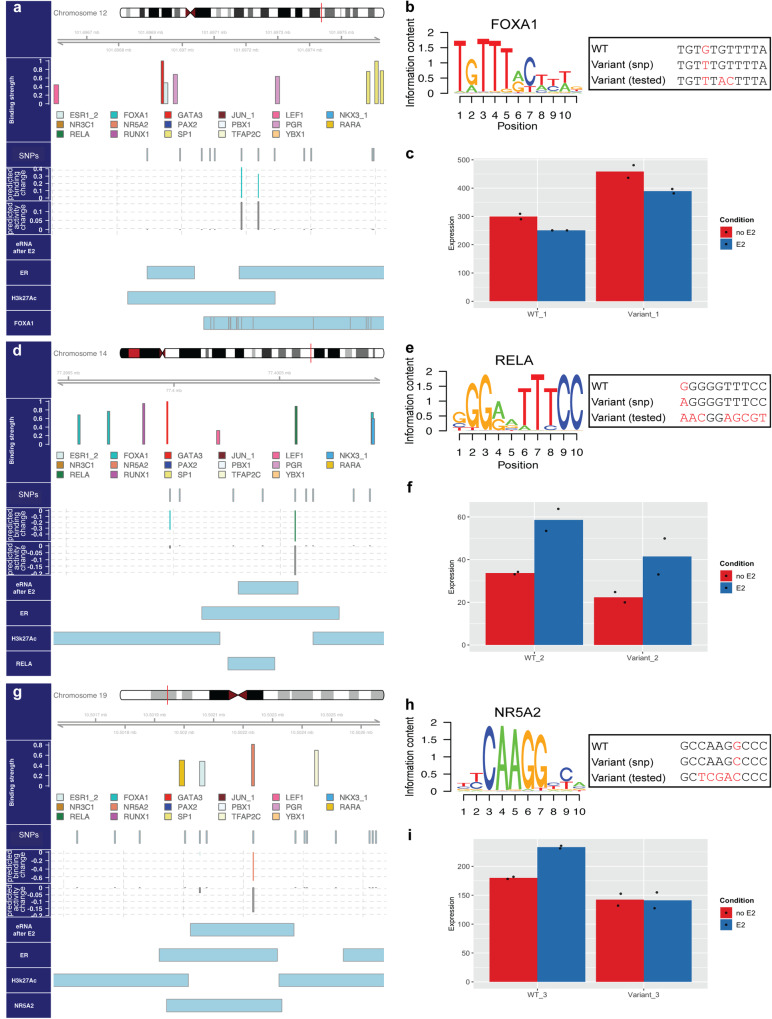
Estrogen Receptor α (ERα) is a major lineage determining transcription factor (TF) in mammary gland development. Dysregulation of ERα-mediated transcriptional program results in cancer. Transcriptomic and epigenomic profiling of breast cancer cell lines has revealed large numbers of enhancers involved in this regulatory program, but how these enhancers encode function in their sequence remains poorly understood. A subset of ERα-bound enhancers are transcribed into short bidirectional RNA (enhancer RNA or eRNA), and this property is believed to be a reliable marker of active enhancers. We therefore analyze thousands of ERα-bound enhancers and build quantitative, mechanism-aware models to discriminate eRNAs from non-transcribing enhancers based on their sequence. Our thermodynamics-based models provide insights into the roles of specific TFs in ERα-mediated transcriptional program, many of which are supported by the literature. We use in silico perturbations to predict TF-enhancer regulatory relationships and integrate these findings with experimentally determined enhancer-promoter interactions to construct a gene regulatory network. We also demonstrate that the model can prioritize breast cancer-related sequence variants while providing mechanistic explanations for their function. Finally, we experimentally validate the model-proposed mechanisms underlying three such variants.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Cooperativity of co-factor NR2F2 with Pioneer Factors GATA3, FOXA1 in promoting ERα function.Theranostics. 2019 Aug 21;9(22):6501-6516. doi: 10.7150/thno.34874. eCollection 2019. Theranostics. 2019. PMID: 31588232 Free PMC article.
-
A hypermethylation strategy utilized by enhancer-bound CARM1 to promote estrogen receptor α-dependent transcriptional activation and breast carcinogenesis.Theranostics. 2020 Feb 10;10(8):3451-3473. doi: 10.7150/thno.39241. eCollection 2020. Theranostics. 2020. PMID: 32206101 Free PMC article.
-
Alternatively Constructed Estrogen Receptor Alpha-Driven Super-Enhancers Result in Similar Gene Expression in Breast and Endometrial Cell Lines.Int J Mol Sci. 2020 Feb 27;21(5):1630. doi: 10.3390/ijms21051630. Int J Mol Sci. 2020. PMID: 32120995 Free PMC article.
-
The first decade of estrogen receptor cistromics in breast cancer.J Endocrinol. 2016 May;229(2):R43-56. doi: 10.1530/JOE-16-0003. Epub 2016 Feb 23. J Endocrinol. 2016. PMID: 26906743 Review.
-
Transcriptional enhancers: functional insights and role in human disease.Curr Opin Genet Dev. 2015 Aug;33:71-6. doi: 10.1016/j.gde.2015.08.009. Epub 2015 Oct 6. Curr Opin Genet Dev. 2015. PMID: 26433090 Free PMC article. Review.
Cited by
-
The role of CDK8 gene polymorphisms in bladder cancer susceptibility and prognosis: a study in the Chinese Han population.BMC Cancer. 2025 Apr 16;25(1):714. doi: 10.1186/s12885-025-14132-w. BMC Cancer. 2025. PMID: 40241036 Free PMC article.
-
Deciphering enhancer sequence using thermodynamics-based models and convolutional neural networks.Nucleic Acids Res. 2021 Oct 11;49(18):10309-10327. doi: 10.1093/nar/gkab765. Nucleic Acids Res. 2021. PMID: 34508359 Free PMC article.
References
-
- Desantis, C. E. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J. Clin.67, 439–448 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R35 GM131819/GM/NIGMS NIH HHS/United States
- R01 GM132458/GM/NIGMS NIH HHS/United States
- R21 AG065748/AG/NIA NIH HHS/United States
- R35GM131819/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- R01GM114341A/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
