

Transcriptomic analysis reveals the regulatory mechanisms of messenger RNA (mRNA) and long non-coding RNA (lncRNA) in response to waterlogging stress in rye (Secale cereale L.)
- PMID: 38862913
- PMCID: PMC11167852
- DOI: 10.1186/s12870-024-05234-x
Transcriptomic analysis reveals the regulatory mechanisms of messenger RNA (mRNA) and long non-coding RNA (lncRNA) in response to waterlogging stress in rye (Secale cereale L.)
Abstract
Background: Waterlogging stress (WS) negatively impacts crop growth and productivity, making it important to understand crop resistance processes and discover useful WS resistance genes. In this study, rye cultivars and wild rye species were subjected to 12-day WS treatment, and the cultivar Secale cereale L. Imperil showed higher tolerance. Whole transcriptome sequencing was performed on this cultivar to identify differentially expressed (DE) messenger RNAs (DE-mRNAs) and long non-coding RNAs (DE-lncRNAs) involved in WS response.
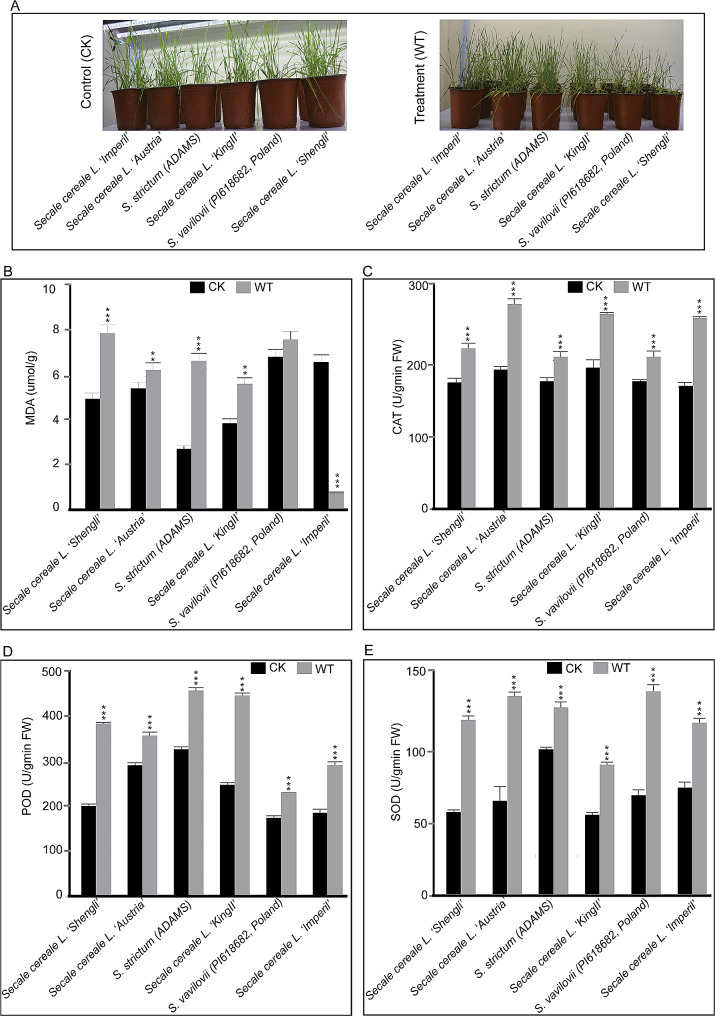
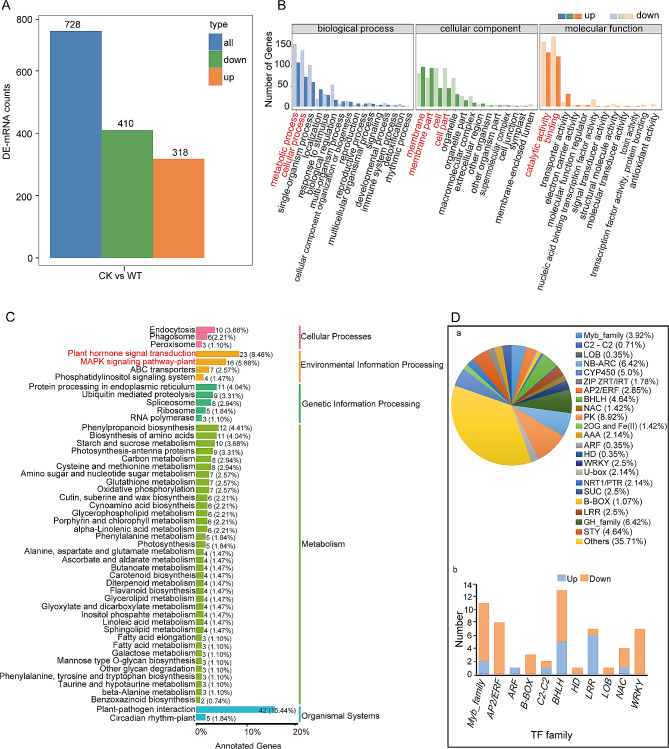
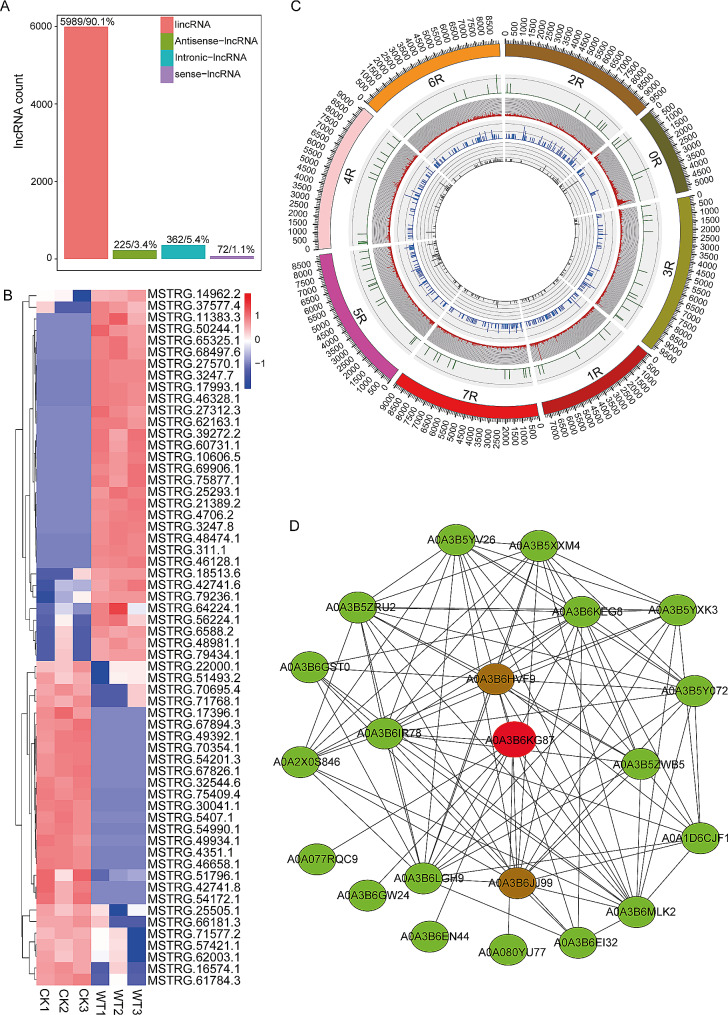
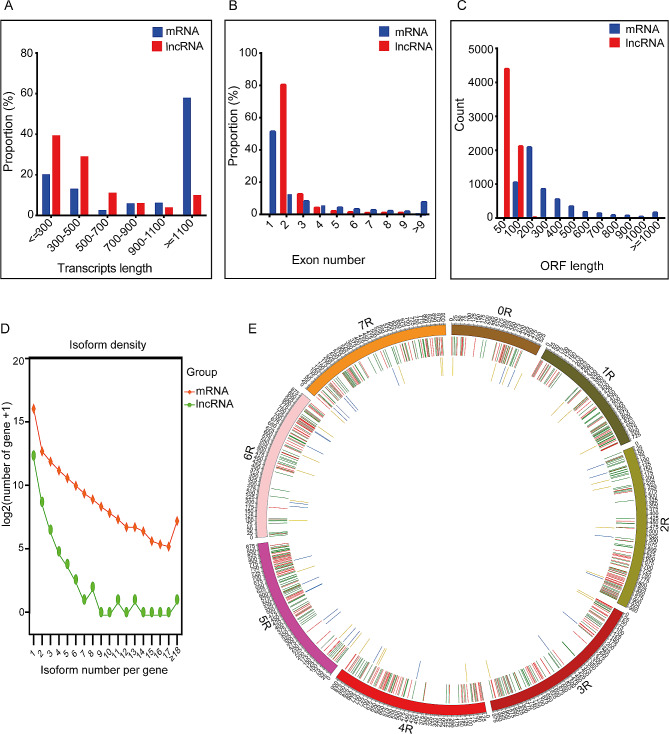
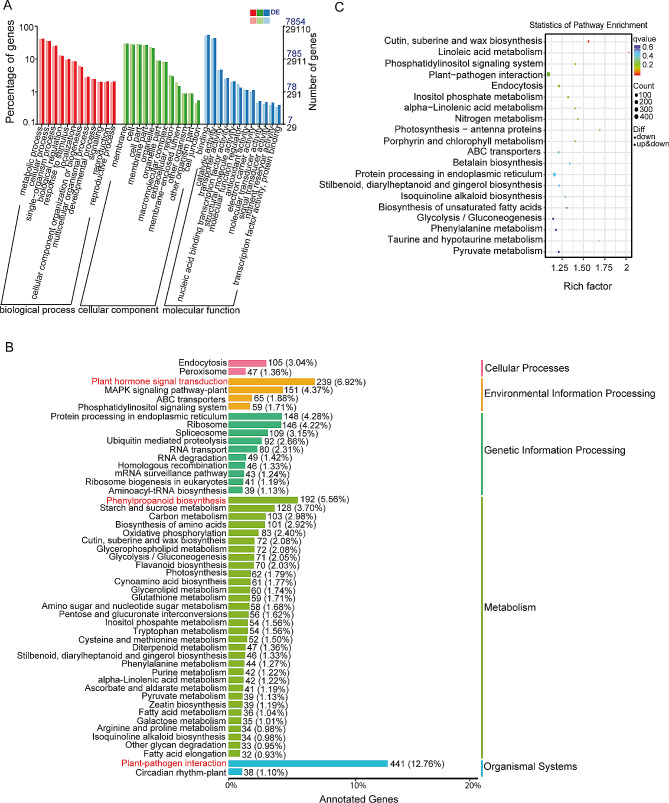
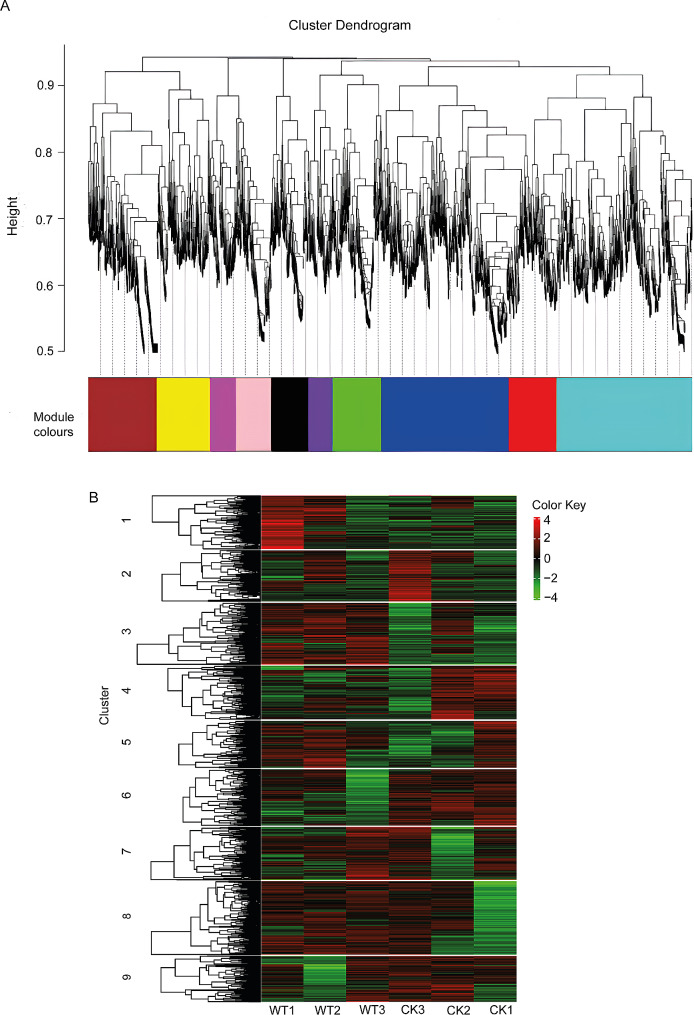
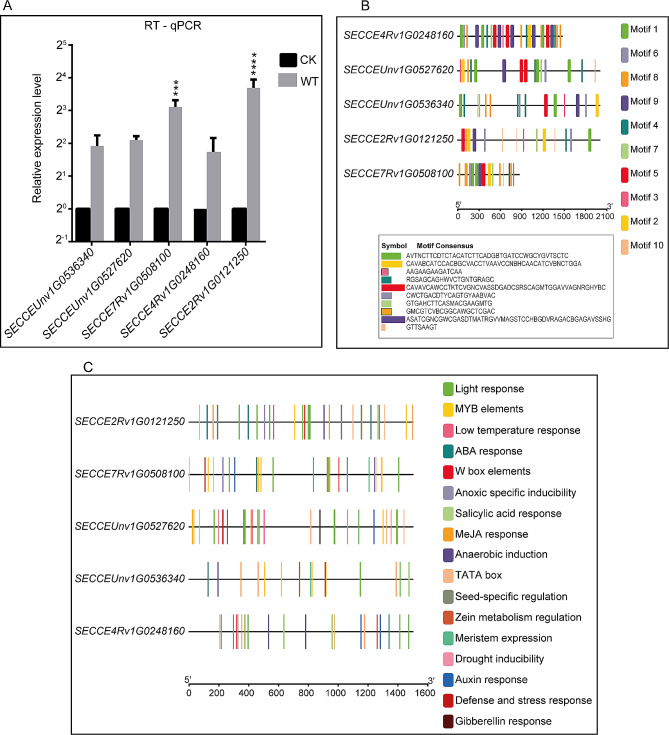
Results: Among the 6 species, Secale cereale L. Imperil showed higher tolerance than wild rye species against WS. The cultivar effectively mitigated oxidative stress, and regulated hydrogen peroxide and superoxide anion. A total of 728 DE-mRNAs and 60 DE-lncRNAs were discovered. Among these, 318 DE-mRNAs and 32 DE-lncRNAs were upregulated, and 410 DE-mRNAs and 28 DE-lncRNAs were downregulated. GO enrichment analysis discovered metabolic processes, cellular processes, and single-organism processes as enriched biological processes (BP). For cellular components (CC), the enriched terms were membrane, membrane part, cell, and cell part. Enriched molecular functions (MF) terms were catalytic activity, binding, and transporter activity. LncRNA and mRNA regulatory processes were mainly related to MAPK signaling pathway-plant, plant hormone signal transduction, phenylpropanoid biosynthesis, anthocyanin biosynthesis, glutathione metabolism, ubiquitin-mediated proteolysis, ABC transporter, Cytochrome b6/f complex, secondary metabolite biosynthesis, and carotenoid biosynthesis pathways. The signalling of ethylene-related pathways was not mainly dependent on AP2/ERF and WRKY transcription factors (TF), but on other factors. Photosynthetic activity was active, and carotenoid levels increased in rye under WS. Sphingolipids, the cytochrome b6/f complex, and glutamate are involved in rye WS response. Sucrose transportation was not significantly inhibited, and sucrose breakdown occurs in rye under WS.
Conclusions: This study investigated the expression levels and regulatory functions of mRNAs and lncRNAs in 12-day waterlogged rye seedlings. The findings shed light on the genes that play a significant role in rye ability to withstand WS. The findings from this study will serve as a foundation for further investigations into the mRNA and lncRNA WS responses in rye.
Keywords: DE-lncRNA; DE-mRNA; Rye (Secale cereale L.); Transcriptome sequencing; Waterlogging stress (WS).
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
A Comprehensive Transcriptomics Analysis Reveals Long Non-Coding RNA to be Involved in the Key Metabolic Pathway in Response to Waterlogging Stress in Maize.Genes (Basel). 2020 Feb 29;11(3):267. doi: 10.3390/genes11030267. Genes (Basel). 2020. PMID: 32121334 Free PMC article.
-
Identification and Network Construction of mRNAs, miRNAs, lncRNAs, and circRNAs in Sweetpotato (Ipomoea batatas L.) Adventitious Roots Under Salt Stress via Whole-Transcriptome RNA Sequencing.Int J Mol Sci. 2025 Feb 15;26(4):1660. doi: 10.3390/ijms26041660. Int J Mol Sci. 2025. PMID: 40004124 Free PMC article.
-
A Comparative Transcriptome Analysis, Conserved Regulatory Elements and Associated Transcription Factors Related to Accumulation of Fusariotoxins in Grain of Rye (Secale cereale L.) Hybrids.Int J Mol Sci. 2020 Oct 8;21(19):7418. doi: 10.3390/ijms21197418. Int J Mol Sci. 2020. PMID: 33049995 Free PMC article.
-
Biological Insights and Recent Advances in Plant Long Non-Coding RNA.Int J Mol Sci. 2024 Nov 7;25(22):11964. doi: 10.3390/ijms252211964. Int J Mol Sci. 2024. PMID: 39596034 Free PMC article. Review.
-
The plant noncoding transcriptome: a versatile environmental sensor.EMBO J. 2023 Oct 16;42(20):e114400. doi: 10.15252/embj.2023114400. Epub 2023 Sep 21. EMBO J. 2023. PMID: 37735935 Free PMC article. Review.
Cited by
-
Epitranscriptomic Control of Drought Tolerance in Rice: The Role of RNA Methylation.Plants (Basel). 2025 Jun 30;14(13):2002. doi: 10.3390/plants14132002. Plants (Basel). 2025. PMID: 40648011 Free PMC article. Review.
-
Evaluating Genome Assemblies for Optimized Completeness and Accuracy of Reference Gene Sequences in Wheat, Rye, and Triticale.Plants (Basel). 2025 Apr 6;14(7):1140. doi: 10.3390/plants14071140. Plants (Basel). 2025. PMID: 40219208 Free PMC article.
-
Physiological and Multi-Omics Integrative Analysis Provides New Insights into Tolerance to Waterlogging Stress in Sesame (Sesamum indicum L.).Int J Mol Sci. 2025 Jan 3;26(1):351. doi: 10.3390/ijms26010351. Int J Mol Sci. 2025. PMID: 39796205 Free PMC article.
References
-
- Islam R, Islam MM, Islam MN, Islam MN, Sen S, Faisal RK. Climate change adaptation strategies: a prospect toward crop modelling and food security management. Model Earth Syst Environ. 2019;6:769–77. doi: 10.1007/s40808-019-00708-6. - DOI
-
- Ding J, Liang P, Wu P, Zhu M, Li C, Zhu X, et al. Effects of waterlogging on grain yield and associated traits of historic wheat cultivars in the middle and lower reaches of the Yangtze River, China. Field Crops Res. 2020;246:107695. doi: 10.1016/j.fcr.2019.107695. - DOI
-
- Mustroph A. Improving flooding tolerance of crop plants. Agronomy. 2018;8(9):160. doi: 10.3390/agronomy8090160. - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
