

Genome-wide enhancer RNA profiling adds molecular links between genetic variation and human cancers
- PMID: 38863031
- PMCID: PMC11165858
- DOI: 10.1186/s40779-024-00539-2
Genome-wide enhancer RNA profiling adds molecular links between genetic variation and human cancers
Abstract
Background: Dysregulation of enhancer transcription occurs in multiple cancers. Enhancer RNAs (eRNAs) are transcribed products from enhancers that play critical roles in transcriptional control. Characterizing the genetic basis of eRNA expression may elucidate the molecular mechanisms underlying cancers.
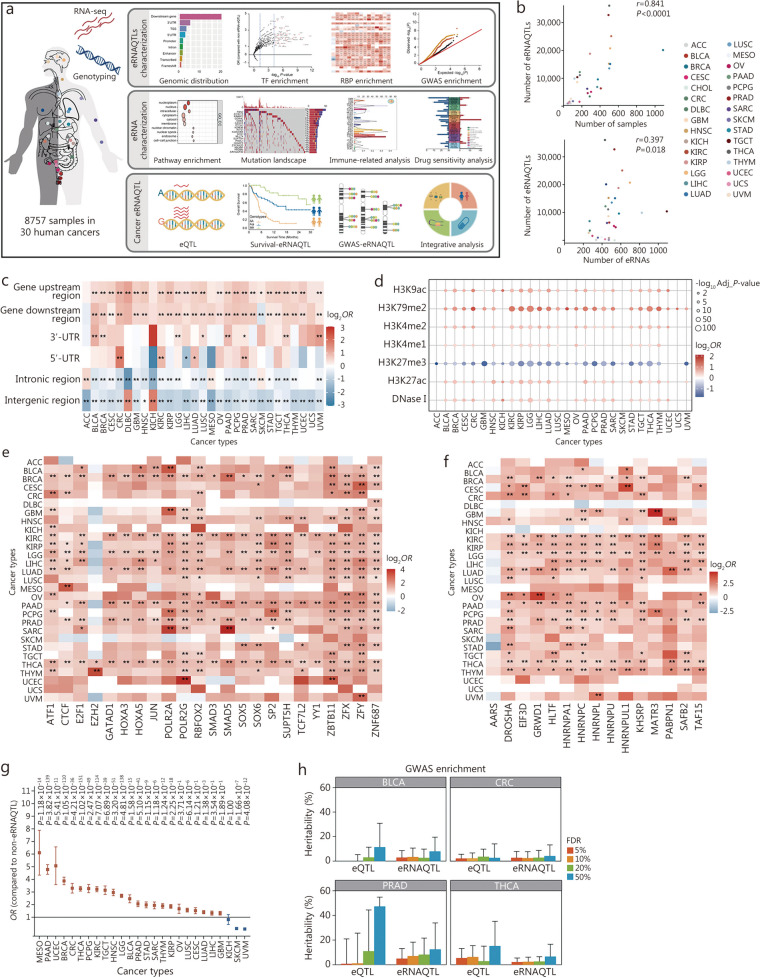
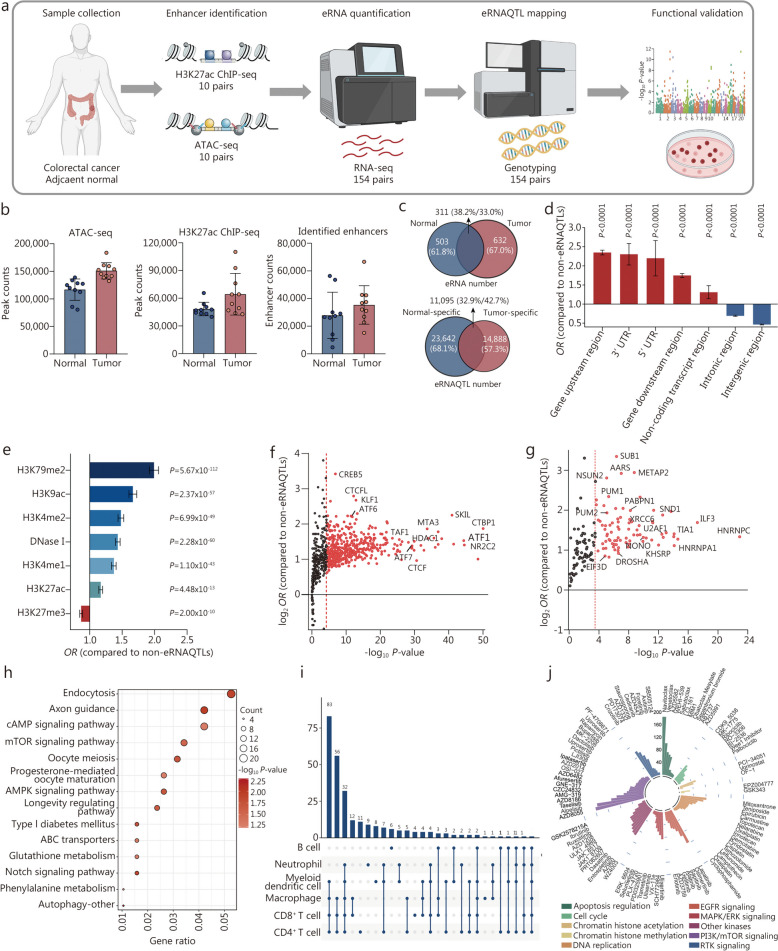
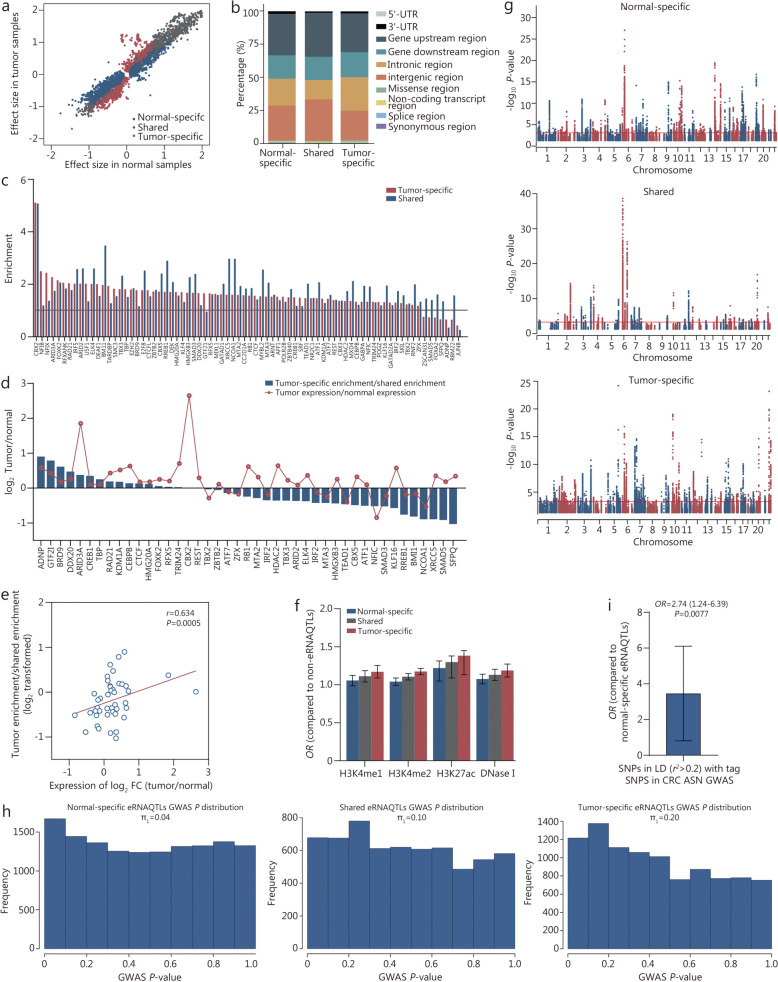
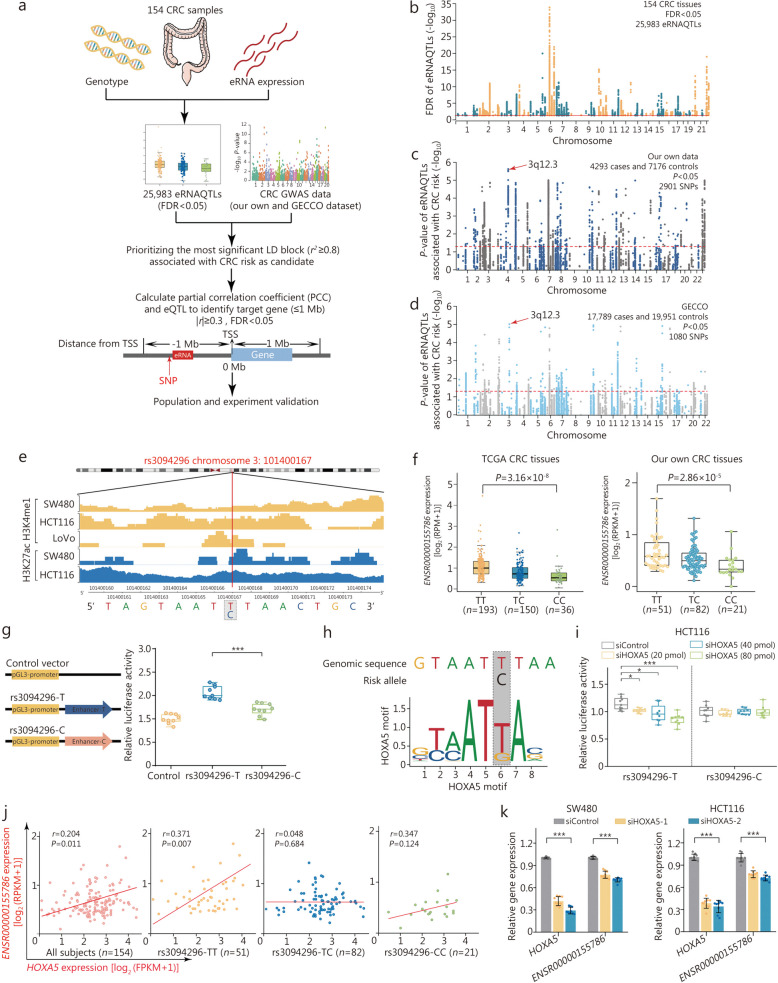
Methods: Initially, a comprehensive analysis of eRNA quantitative trait loci (eRNAQTLs) was performed in The Cancer Genome Atlas (TCGA), and functional features were characterized using multi-omics data. To establish the first eRNAQTL profiles for colorectal cancer (CRC) in China, epigenomic data were used to define active enhancers, which were subsequently integrated with transcription and genotyping data from 154 paired CRC samples. Finally, large-scale case-control studies (34,585 cases and 69,544 controls) were conducted along with multipronged experiments to investigate the potential mechanisms by which candidate eRNAQTLs affect CRC risk.
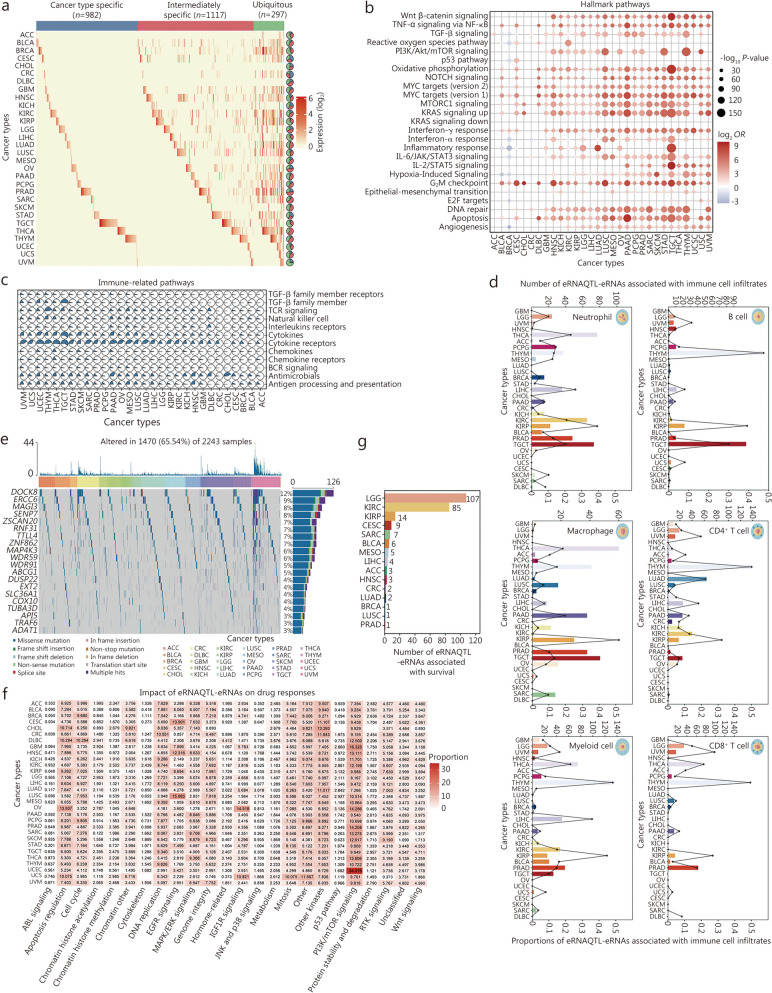
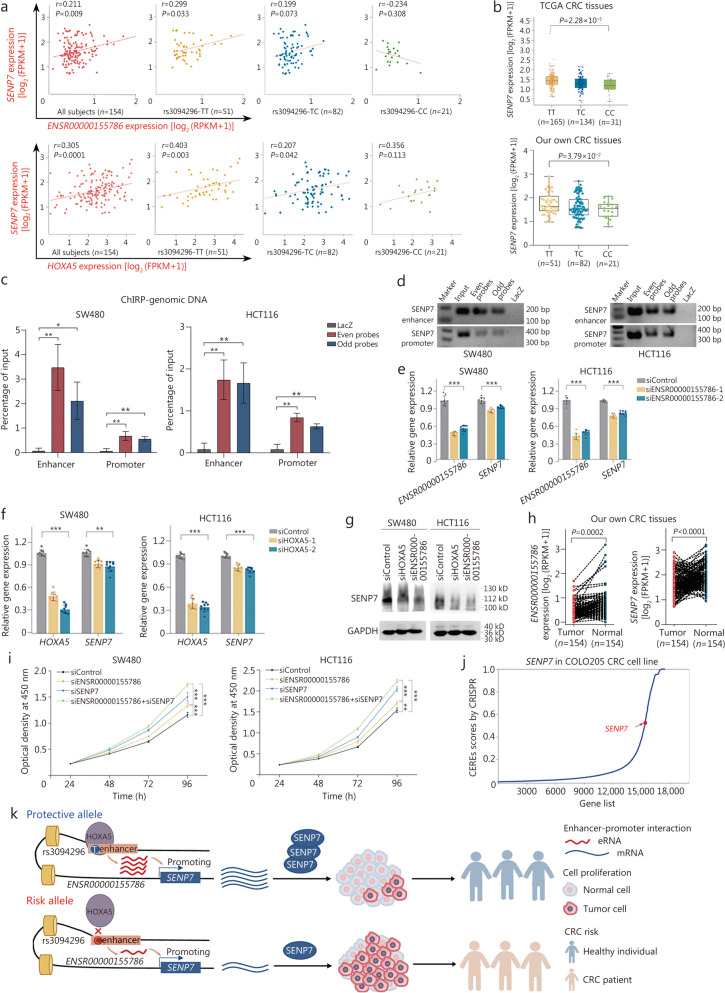
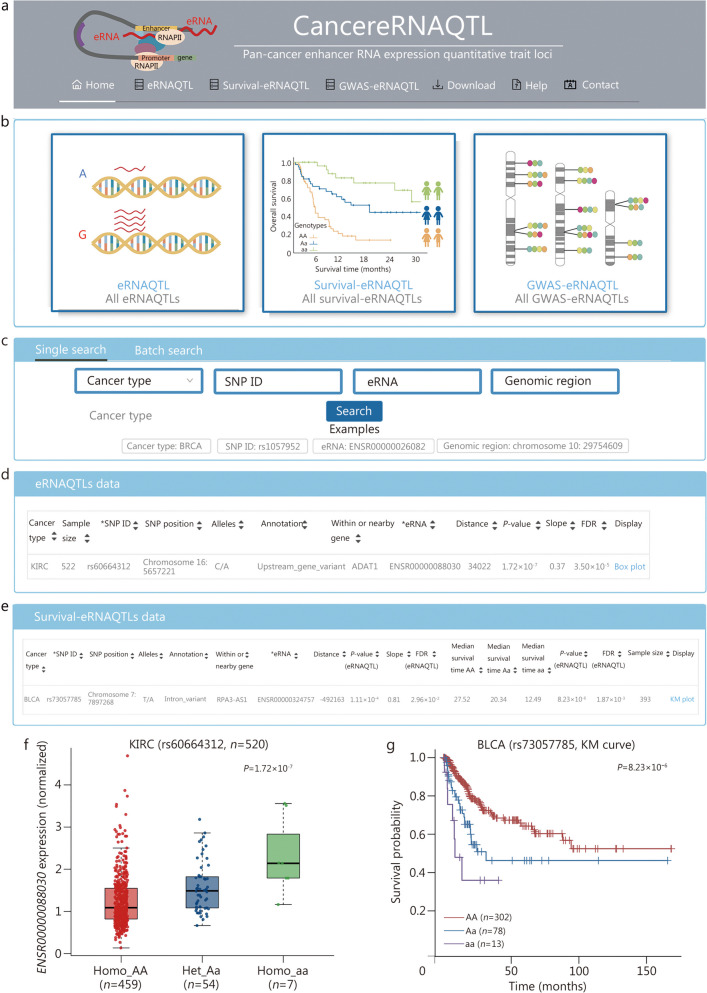
Results: A total of 300,112 eRNAQTLs were identified across 30 different cancer types, which exert their influence on eRNA transcription by modulating chromatin status, binding affinity to transcription factors and RNA-binding proteins. These eRNAQTLs were found to be significantly enriched in cancer risk loci, explaining a substantial proportion of cancer heritability. Additionally, tumor-specific eRNAQTLs exhibited high responsiveness to the development of cancer. Moreover, the target genes of these eRNAs were associated with dysregulated signaling pathways and immune cell infiltration in cancer, highlighting their potential as therapeutic targets. Furthermore, multiple ethnic population studies have confirmed that an eRNAQTL rs3094296-T variant decreases the risk of CRC in populations from China (OR = 0.91, 95%CI 0.88-0.95, P = 2.92 × 10-7) and Europe (OR = 0.92, 95%CI 0.88-0.95, P = 4.61 × 10-6). Mechanistically, rs3094296 had an allele-specific effect on the transcription of the eRNA ENSR00000155786, which functioned as a transcriptional activator promoting the expression of its target gene SENP7. These two genes synergistically suppressed tumor cell proliferation. Our curated list of variants, genes, and drugs has been made available in CancereRNAQTL ( http://canernaqtl.whu.edu.cn/#/ ) to serve as an informative resource for advancing this field.
Conclusion: Our findings underscore the significance of eRNAQTLs in transcriptional regulation and disease heritability, pinpointing the potential of eRNA-based therapeutic strategies in cancers.
Keywords: ENSR00000155786; SENP7; Enhancer RNA; Genome-wide association study (GWAS); eRNA quantitative trait loci (eRNAQTLs).
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
- NSFC-82103929/The Program of National Natural Science Foundation of China
- NSFC-82273713/The Program of National Natural Science Foundation of China
- 2022QNRC001/Young Elite Scientists Sponsorship Program by CAST
- NSFC-81925032/Distinguished Young Scholars of China
- NSFC-82130098/Key Program of National Natural Science Foundation of China
- 2042022rc0026/Fundamental Research Funds for the Central Universities
- 2042023kf1005/Fundamental Research Funds for the Central Universities
- WHU:2042022kf1205/Fundamental Research Funds for the Central Universities
- WHU: 2042022kf1031/Fundamental Research Funds for the Central Universities
- whkxjsj011/Knowledge Innovation Program of Wuhan
- 2023020201010073/Knowledge Innovation Program of Wuhan
- NSFC-82003547/Youth Program of National Natural Science Foundation of China
- WJ2023M045/Program of Health Commission of Hubei Province
- NSFC-82322058/National Science Fund for Excellent Young Scholars
- 2023AFA046/National Science Fund for Distinguished Young Scholars of Hubei Province of China
- znpy2019060/Science and Technology Innovation Seed Fund of Zhongnan Hospital of Wuhan University
LinkOut - more resources
Full Text Sources
Medical
