

Disease spectrum, prevalence, genetic characteristics of inborn errors of metabolism in 21,840 hospitalized infants in Chongqing, China, 2017-2022
- PMID: 38863445
- PMCID: PMC11165094
- DOI: 10.3389/fgene.2024.1395988
Disease spectrum, prevalence, genetic characteristics of inborn errors of metabolism in 21,840 hospitalized infants in Chongqing, China, 2017-2022
Erratum in
-
Corrigendum: Disease spectrum, prevalence, genetic characteristics of inborn errors of metabolism in 21,840 hospitalized infants in Chongqing, China, 2017-2022.Front Genet. 2024 Jul 10;15:1449534. doi: 10.3389/fgene.2024.1449534. eCollection 2024. Front Genet. 2024. PMID: 39050252 Free PMC article.
Abstract
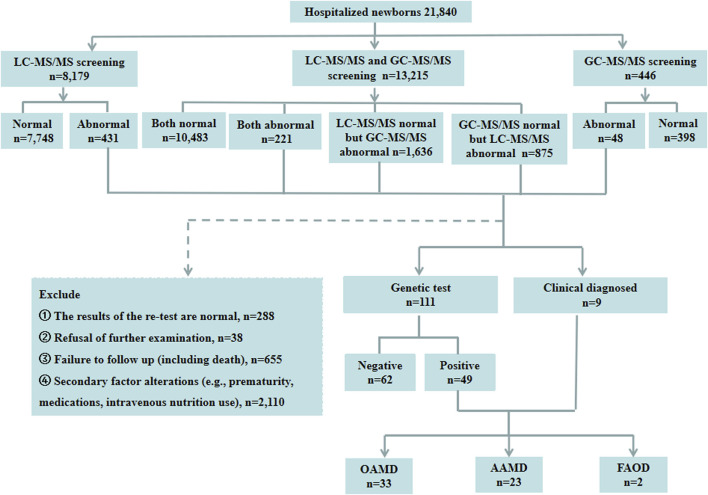
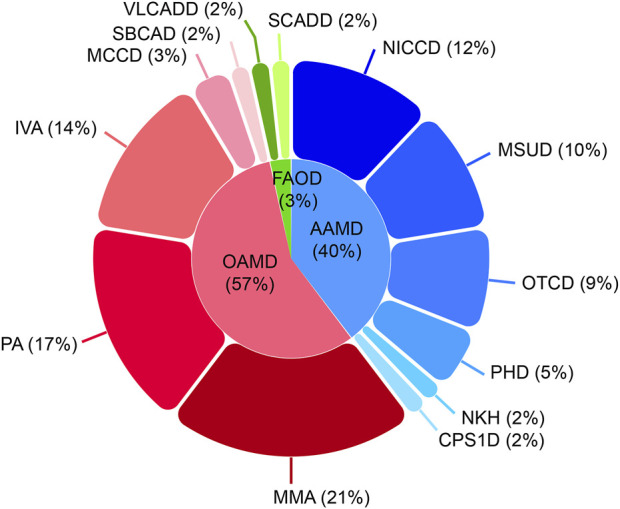
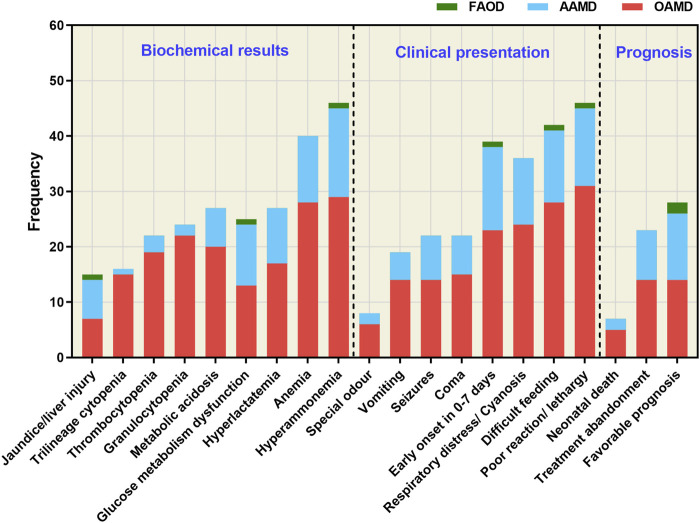
Inborn errors of metabolism (IEMs) are uncommon. Although some studies have explored the distribution and characteristics of IEMs in newborns, the impact of these disorders on hospitalized newborns remains unclear. In this study, we gathered data from 21,840 newborn patients admitted for various medical conditions at the Children's Hospital of Chongqing Medical University from January 2017 and December 2022. Liquid chromatography-tandem mass spectrometry (LC-MS/MS), gas chromatography-mass spectrometry (GC-MS/MS), and genetic analysis were used to elucidate the disease spectrum, incidence rate, and genetic characteristics of IEMs in hospitalized newborns. The results revealed that the incidence of IEMs in hospitalized newborns was 1/377 (58/21,840), with a higher incidence in full-term infants (1/428) than in premature infants (1/3,120). Among the diagnosed genetic metabolic diseases, organic acid metabolism disorders (1/662), amino acid metabolism disorders (1/950), and fatty acid oxidation disorders (1/10,920) were the most prevalent. Methylmalonic acidemia (MMA), especially the isolated form, emerged as the most common IEM, while neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) and ornithine transcarbamylase deficiency (OTCD) were prevalent in premature infants. Of the 58 confirmed cases of IEMs, 72 variants were identified, of which 31.94% (23/72) had not been reported previously. This study contributes to understanding the incidence and clinical features of IEMs in hospitalized newborns, offering more efficient strategies for screening and diagnosing these disorders.
Keywords: disease spectrum; genetic characteristics; inborn errors of metabolism; newborn screening; tandem mass spectrometry.
Copyright © 2024 Wang, Zhang, Yang, Zhang, Wang, Yu, Yang, Huang, Liu, Tang and He.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Screening of 1.17 million newborns for inborn errors of metabolism using tandem mass spectrometry in Shanghai, China: A 19-year report.Mol Genet Metab. 2024 Jan;141(1):108098. doi: 10.1016/j.ymgme.2023.108098. Epub 2023 Nov 30. Mol Genet Metab. 2024. PMID: 38061323
-
Newborn screening for inborn errors of metabolism in a northern Chinese population.J Pediatr Endocrinol Metab. 2023 Jan 20;36(3):278-282. doi: 10.1515/jpem-2022-0543. Print 2023 Mar 28. J Pediatr Endocrinol Metab. 2023. PMID: 36662638
-
Expanded Newborn Screening for Inborn Errors of Metabolism by Tandem Mass Spectrometry in Suzhou, China: Disease Spectrum, Prevalence, Genetic Characteristics in a Chinese Population.Front Genet. 2019 Oct 29;10:1052. doi: 10.3389/fgene.2019.01052. eCollection 2019. Front Genet. 2019. PMID: 31737040 Free PMC article.
-
Aminoacidopathies: Prevalence, Etiology, Screening, and Treatment Options.Biochem Genet. 2018 Apr;56(1-2):7-21. doi: 10.1007/s10528-017-9825-6. Epub 2017 Nov 1. Biochem Genet. 2018. PMID: 29094226 Review.
-
Applications of quantitative metabolomics to revolutionize early diagnosis of inborn errors of metabolism in India.Anal Sci Adv. 2021 Aug 5;2(11-12):546-563. doi: 10.1002/ansa.202100010. eCollection 2021 Dec. Anal Sci Adv. 2021. PMID: 38715861 Free PMC article. Review.
References
-
- Almási T., Guey L. T., Lukacs C., Csetneki K., Vokó Z., Zelei T. (2019). Systematic literature review and meta-analysis on the epidemiology of methylmalonic acidemia (MMA) with a focus on MMA caused by methylmalonyl-CoA mutase (mut) deficiency. Orphanet J. Rare Dis. 14 (1), 84. 10.1186/s13023-019-1063-z - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous