

BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA
- PMID: 38866550
- PMCID: PMC11216308
- DOI: 10.1101/gr.278090.123
BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA
Abstract
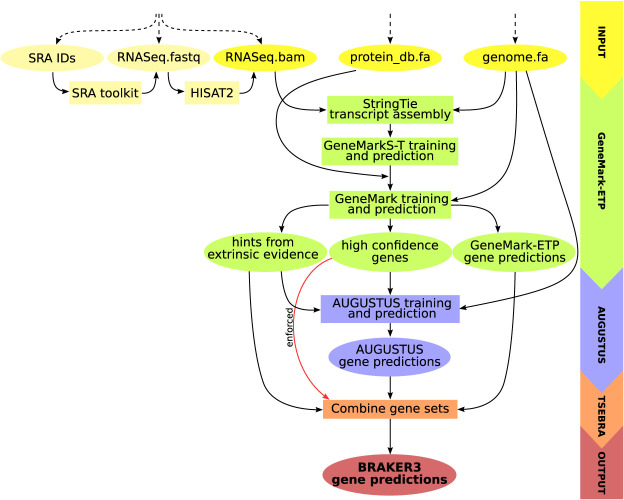
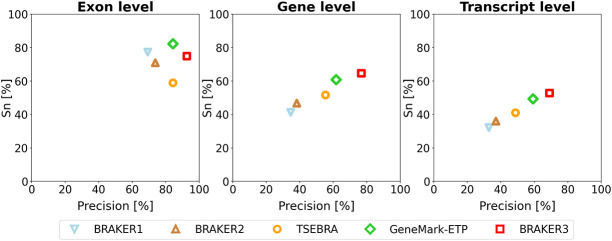
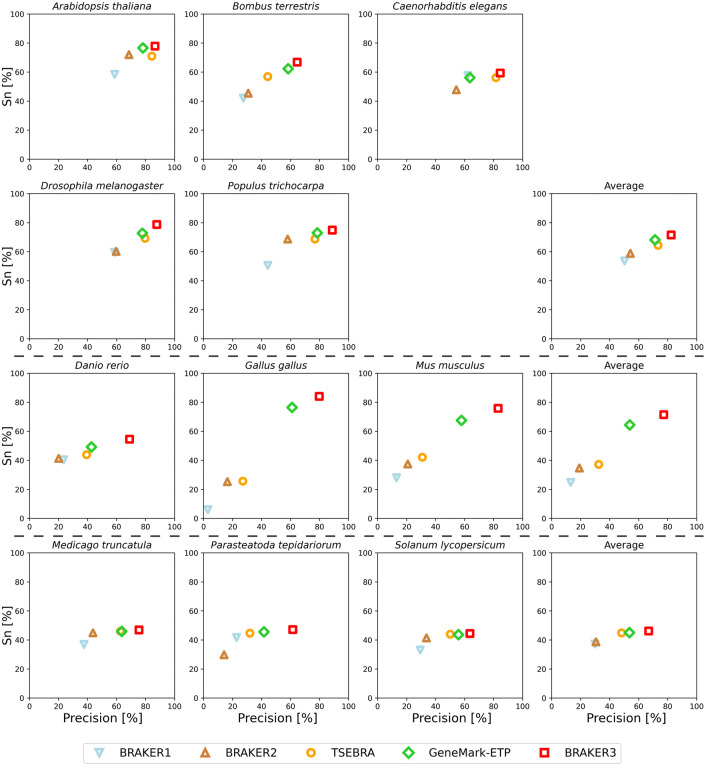
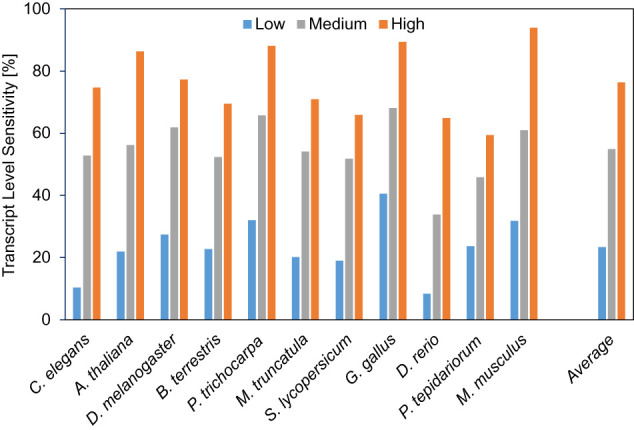
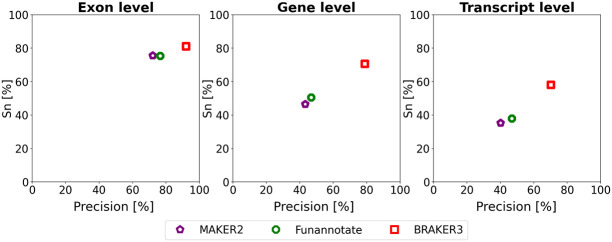
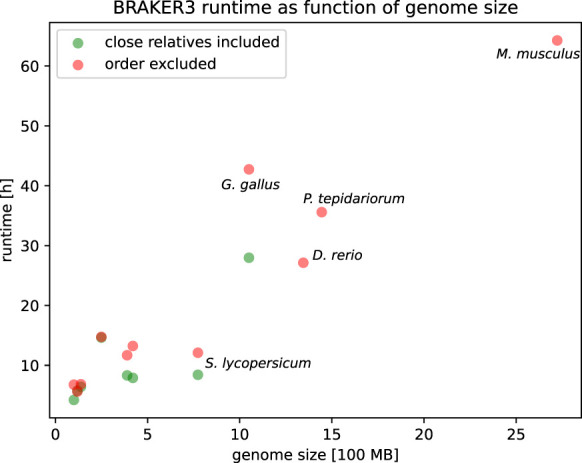
Gene prediction has remained an active area of bioinformatics research for a long time. Still, gene prediction in large eukaryotic genomes presents a challenge that must be addressed by new algorithms. The amount and significance of the evidence available from transcriptomes and proteomes vary across genomes, between genes, and even along a single gene. User-friendly and accurate annotation pipelines that can cope with such data heterogeneity are needed. The previously developed annotation pipelines BRAKER1 and BRAKER2 use RNA-seq or protein data, respectively, but not both. A further significant performance improvement integrating all three data types was made by the recently released GeneMark-ETP. We here present the BRAKER3 pipeline that builds on GeneMark-ETP and AUGUSTUS, and further improves accuracy using the TSEBRA combiner. BRAKER3 annotates protein-coding genes in eukaryotic genomes using both short-read RNA-seq and a large protein database, along with statistical models learned iteratively and specifically for the target genome. We benchmarked the new pipeline on genomes of 11 species under an assumed level of relatedness of the target species proteome to available proteomes. BRAKER3 outperforms BRAKER1 and BRAKER2. The average transcript-level F1-score is increased by about 20 percentage points on average, whereas the difference is most pronounced for species with large and complex genomes. BRAKER3 also outperforms other existing tools, MAKER2, Funannotate, and FINDER. The code of BRAKER3 is available on GitHub and as a ready-to-run Docker container for execution with Docker or Singularity. Overall, BRAKER3 is an accurate, easy-to-use tool for eukaryotic genome annotation.
© 2024 Gabriel et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Update of
-
BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA.bioRxiv [Preprint]. 2024 Feb 29:2023.06.10.544449. doi: 10.1101/2023.06.10.544449. bioRxiv. 2024. Update in: Genome Res. 2024 Jun 25;34(5):769-777. doi: 10.1101/gr.278090.123. PMID: 37398387 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources