

Entire expressed peripheral blood transcriptome in pediatric severe malarial anemia
- PMID: 38866743
- PMCID: PMC11169501
- DOI: 10.1038/s41467-024-48259-4
Entire expressed peripheral blood transcriptome in pediatric severe malarial anemia
Abstract
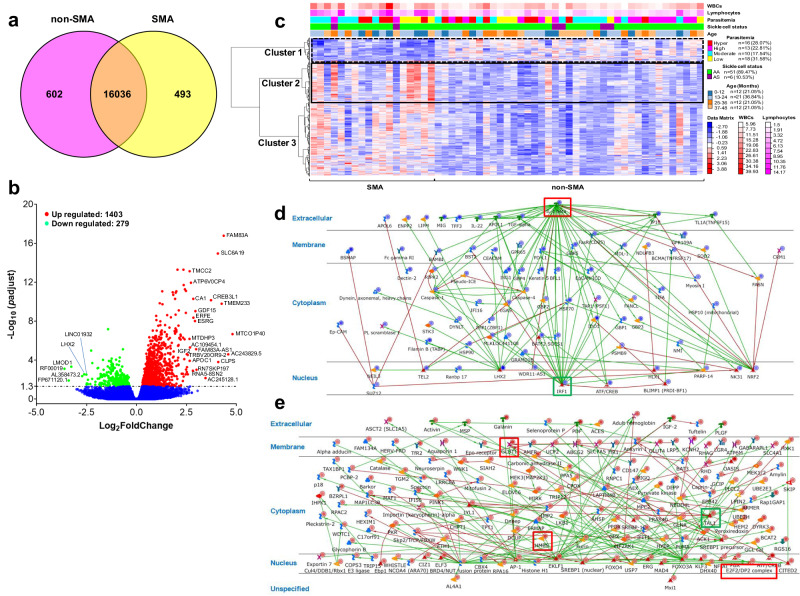
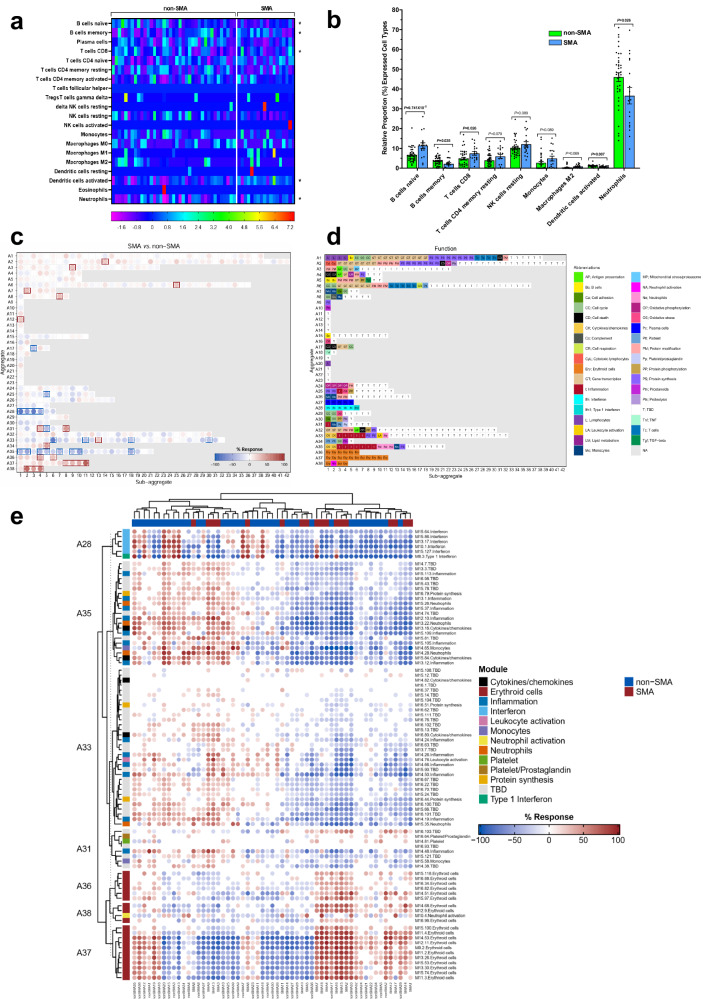
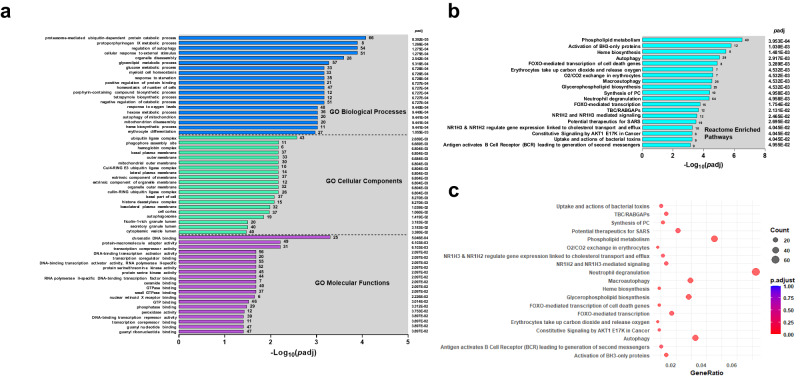
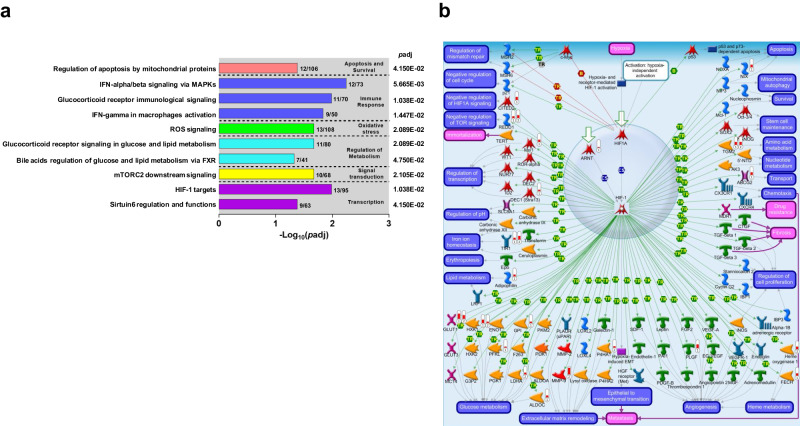
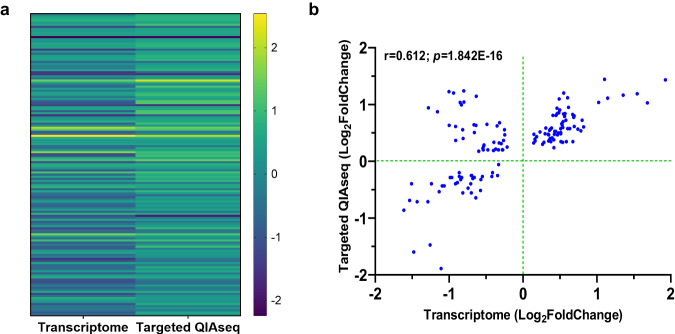
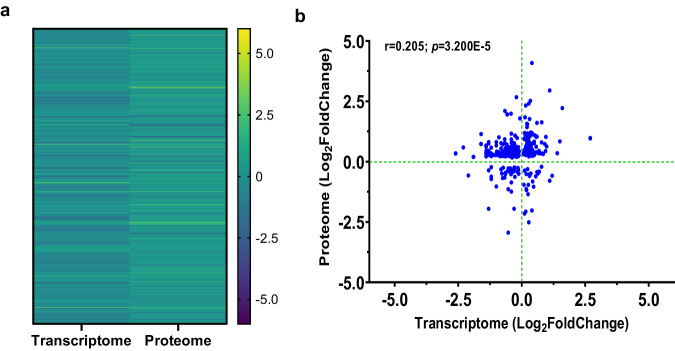
This study on severe malarial anemia (SMA: Hb < 6.0 g/dL), a leading global cause of childhood morbidity and mortality, compares the entire expressed whole blood host transcriptome between Kenyan children (3-48 mos.) with non-SMA (Hb ≥ 6.0 g/dL, n = 39) and SMA (n = 18). Differential expression analyses reveal 1403 up-regulated and 279 down-regulated transcripts in SMA, signifying impairments in host inflammasome activation, cell death, and innate immune and cellular stress responses. Immune cell profiling shows decreased memory responses, antigen presentation, and immediate pathogen clearance, suggesting an immature/improperly regulated immune response in SMA. Module repertoire analysis of blood-specific gene signatures identifies up-regulation of erythroid genes, enhanced neutrophil activation, and impaired inflammatory responses in SMA. Enrichment analyses converge on disruptions in cellular homeostasis and regulatory pathways for the ubiquitin-proteasome system, autophagy, and heme metabolism. Pathway analyses highlight activation in response to hypoxic conditions [Hypoxia Inducible Factor (HIF)-1 target and Reactive Oxygen Species (ROS) signaling] as a central theme in SMA. These signaling pathways are also top-ranking in protein abundance measures and a Ugandan SMA cohort with available transcriptomic data. Targeted RNA-Seq validation shows strong concordance with our entire expressed transcriptome data. These findings identify key molecular themes in SMA pathogenesis, offering potential targets for new malaria therapies.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Entire Expressed Peripheral Blood Transcriptome in Pediatric Severe Malarial Anemia.Res Sq [Preprint]. 2023 Jul 19:rs.3.rs-3150748. doi: 10.21203/rs.3.rs-3150748/v1. Res Sq. 2023. Update in: Nat Commun. 2024 Jun 12;15(1):5037. doi: 10.1038/s41467-024-48259-4. PMID: 37503086 Free PMC article. Updated. Preprint.
References
-
- WHO. World Malaria Report 2023. Report No. CC BY-NC-SA 3.0 IGO, (Geneva, 2023).
MeSH terms
Substances
Grants and funding
- 20150090DR/DOE | LDRD | Los Alamos National Laboratory (Los Alamos Lab)
- D43 TW005884/TW/FIC NIH HHS/United States
- D43TW05884/U.S. Department of Health & Human Services | NIH | Fogarty International Center (FIC)
- D43TW010543/U.S. Department of Health & Human Services | NIH | Fogarty International Center (FIC)
- R01AI130473/Division of Intramural Research, National Institute of Allergy and Infectious Diseases (Division of Intramural Research of the NIAID)
- K43 TW011581/TW/FIC NIH HHS/United States
- K43TW011581/U.S. Department of Health & Human Services | NIH | Fogarty International Center (FIC)
- D43 TW010543/TW/FIC NIH HHS/United States
- R01AI51305/Division of Intramural Research, National Institute of Allergy and Infectious Diseases (Division of Intramural Research of the NIAID)
- R01 AI051305/AI/NIAID NIH HHS/United States
- R01 AI130473/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
