Strand-resolved mutagenicity of DNA damage and repair
- PMID: 38867042
- PMCID: PMC11186772
- DOI: 10.1038/s41586-024-07490-1
Strand-resolved mutagenicity of DNA damage and repair
Abstract
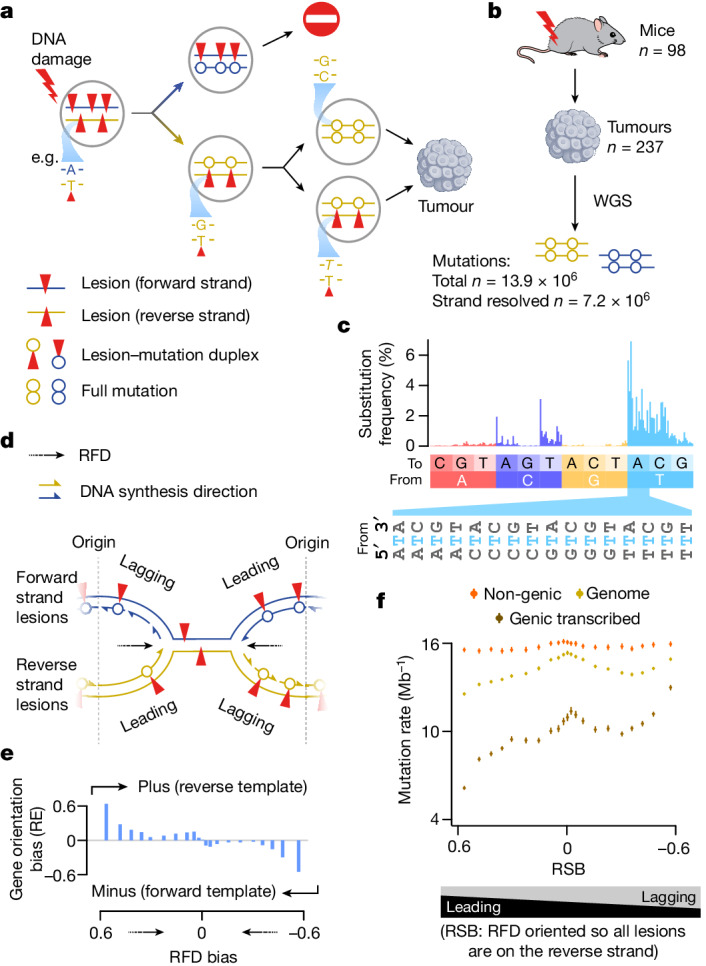
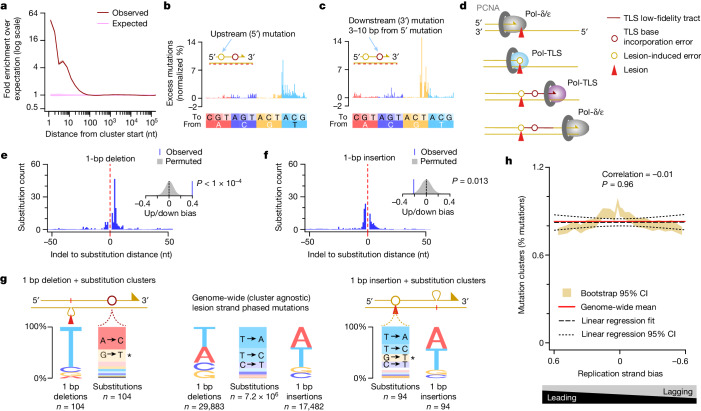
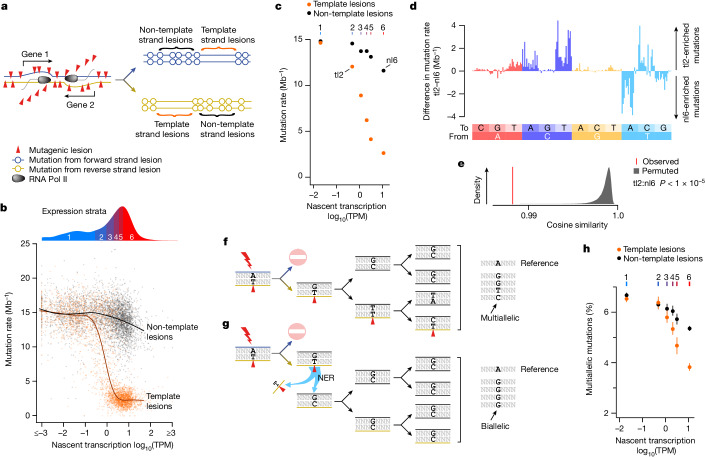
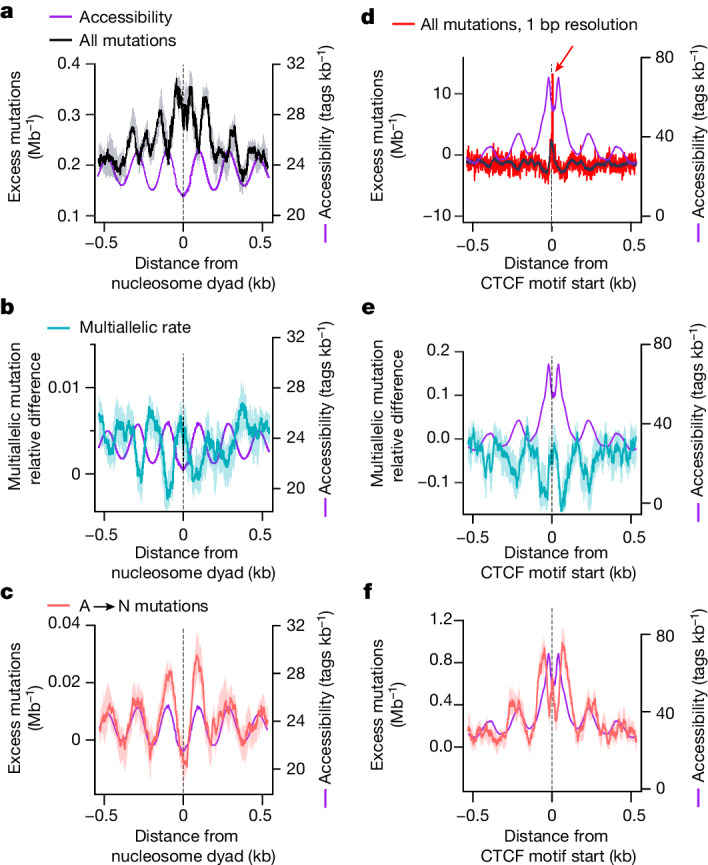
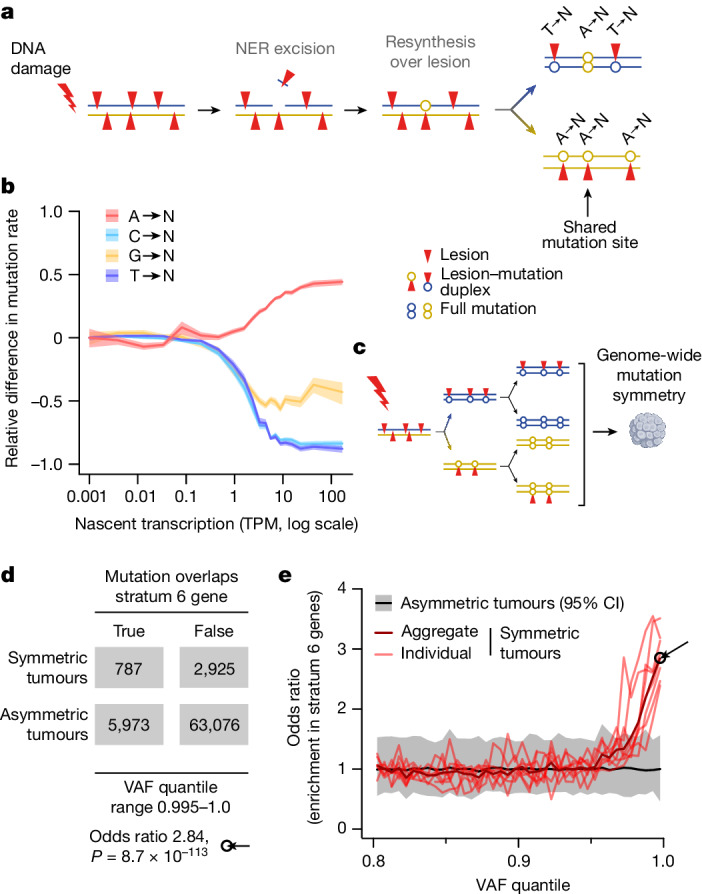
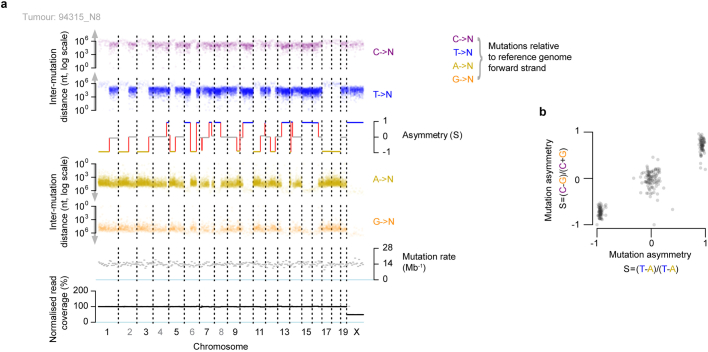
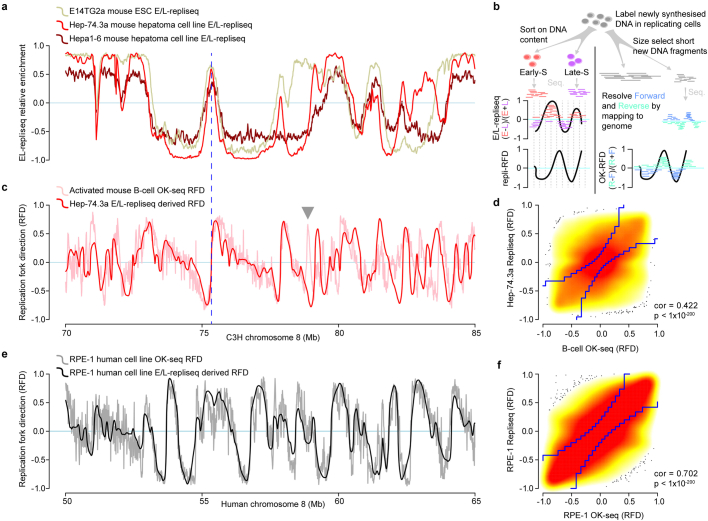
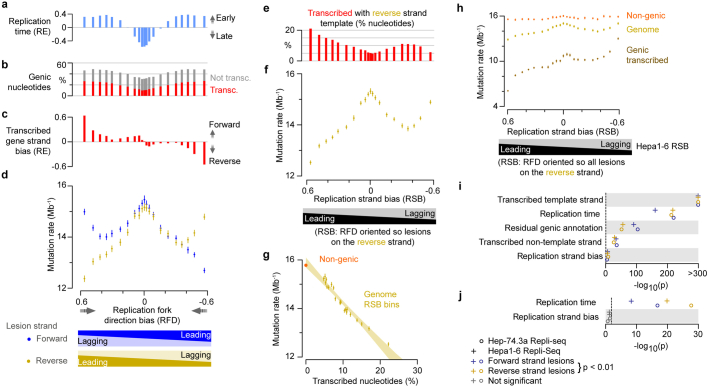
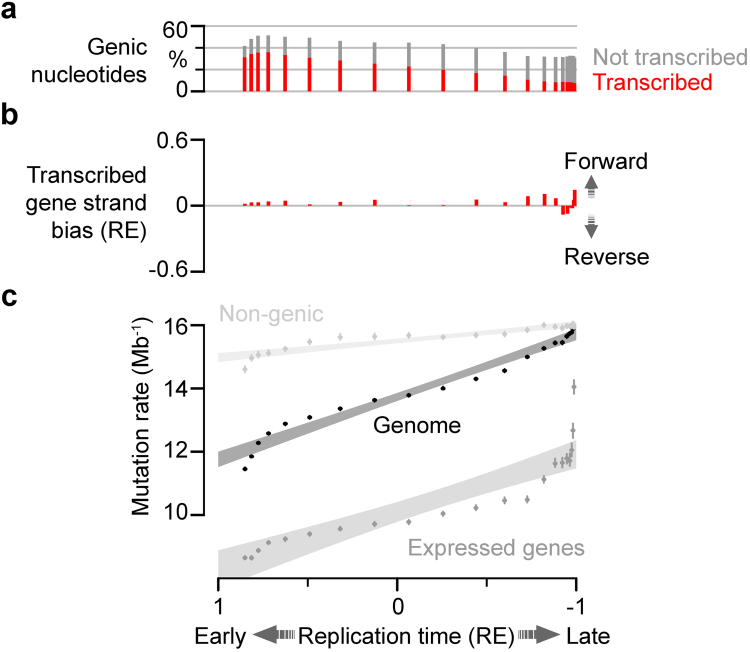
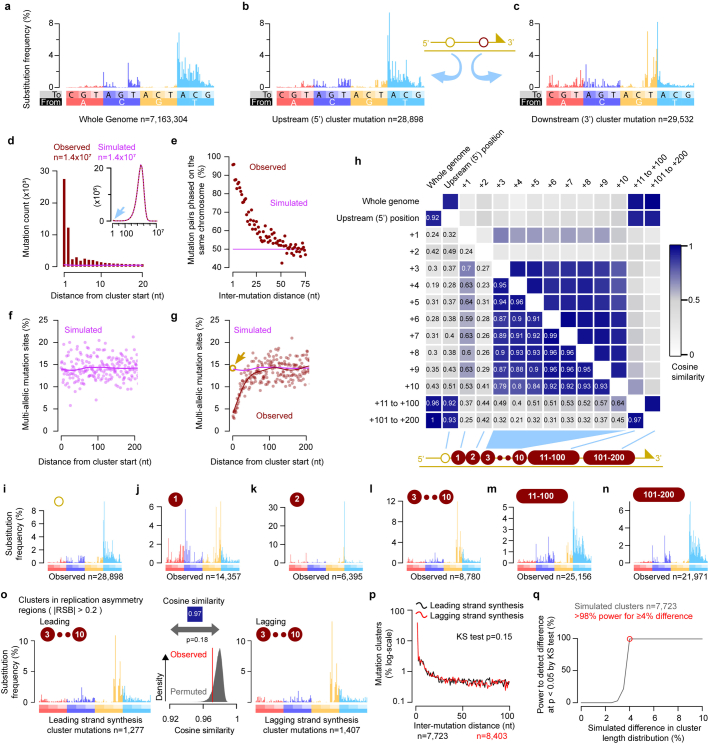
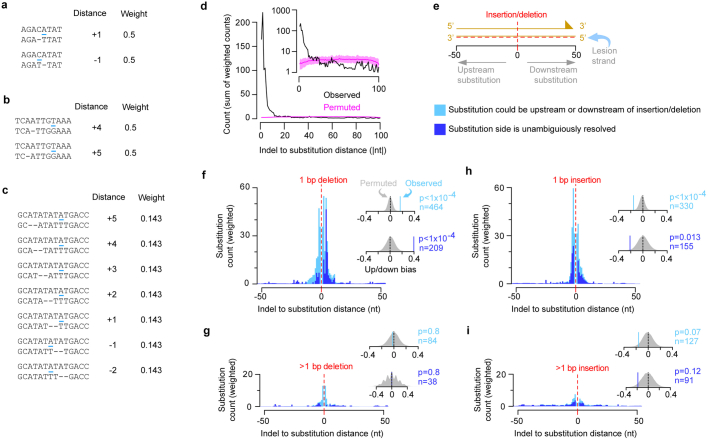
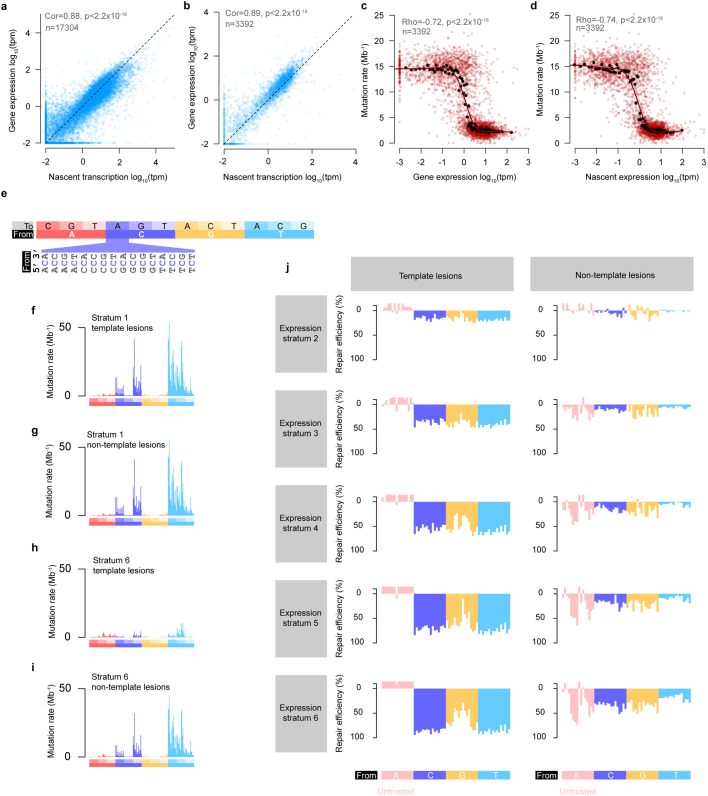
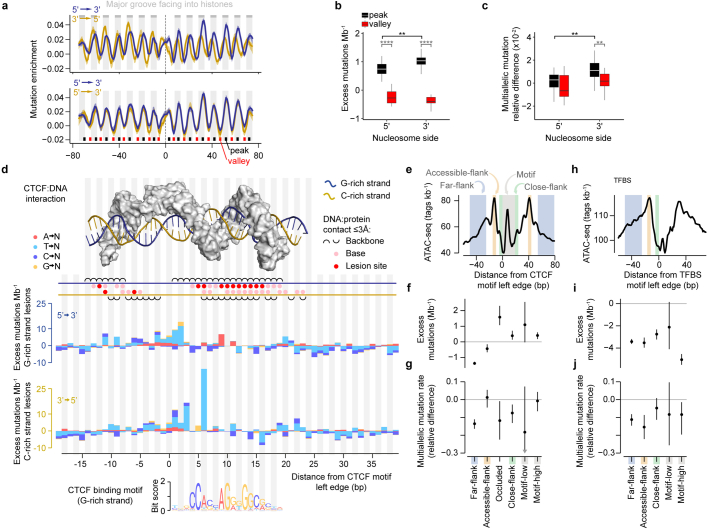
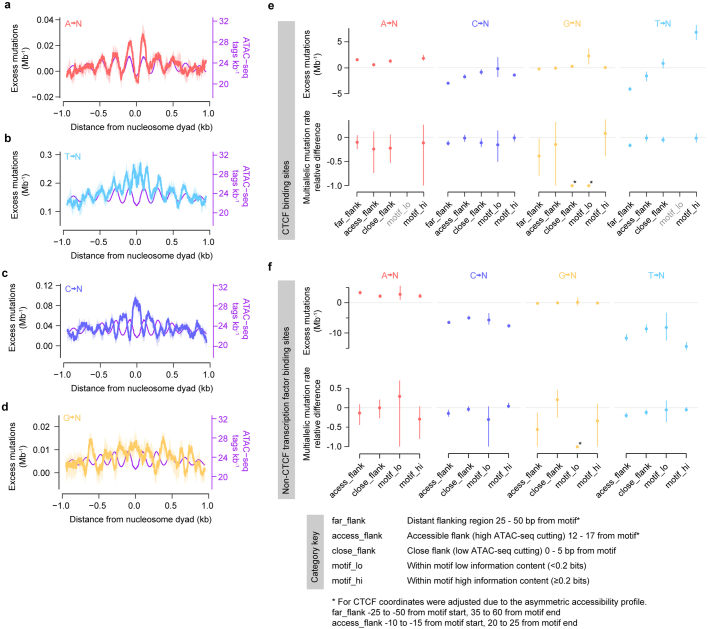
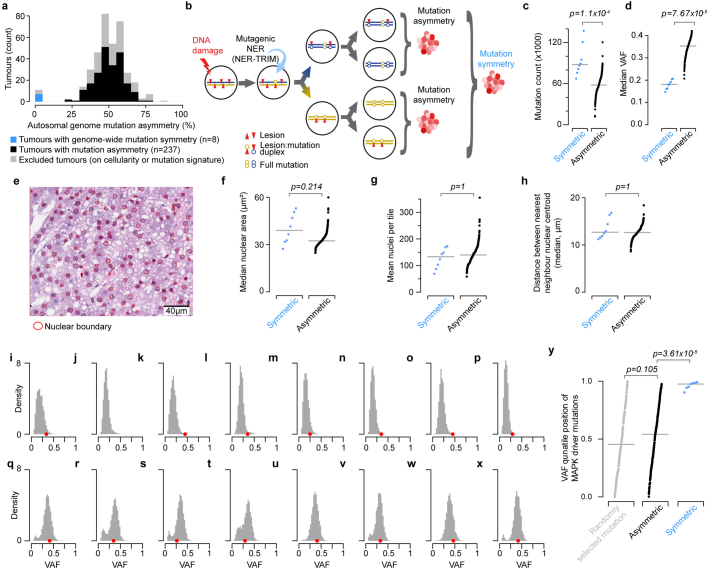
DNA base damage is a major source of oncogenic mutations1. Such damage can produce strand-phased mutation patterns and multiallelic variation through the process of lesion segregation2. Here we exploited these properties to reveal how strand-asymmetric processes, such as replication and transcription, shape DNA damage and repair. Despite distinct mechanisms of leading and lagging strand replication3,4, we observe identical fidelity and damage tolerance for both strands. For small alkylation adducts of DNA, our results support a model in which the same translesion polymerase is recruited on-the-fly to both replication strands, starkly contrasting the strand asymmetric tolerance of bulky UV-induced adducts5. The accumulation of multiple distinct mutations at the site of persistent lesions provides the means to quantify the relative efficiency of repair processes genome wide and at single-base resolution. At multiple scales, we show DNA damage-induced mutations are largely shaped by the influence of DNA accessibility on repair efficiency, rather than gradients of DNA damage. Finally, we reveal specific genomic conditions that can actively drive oncogenic mutagenesis by corrupting the fidelity of nucleotide excision repair. These results provide insight into how strand-asymmetric mechanisms underlie the formation, tolerance and repair of DNA damage, thereby shaping cancer genome evolution.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures