

Ancient Plasmodium genomes shed light on the history of human malaria
- PMID: 38867050
- PMCID: PMC11222158
- DOI: 10.1038/s41586-024-07546-2
Ancient Plasmodium genomes shed light on the history of human malaria
Abstract
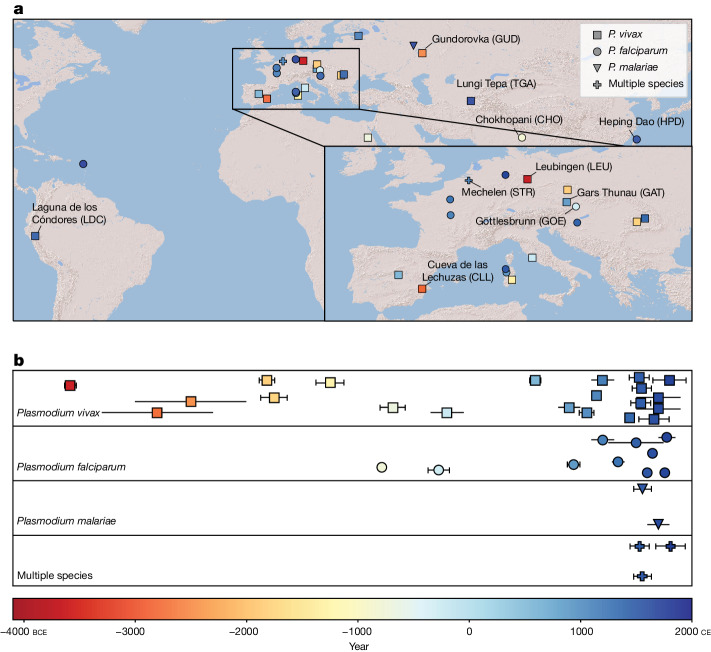
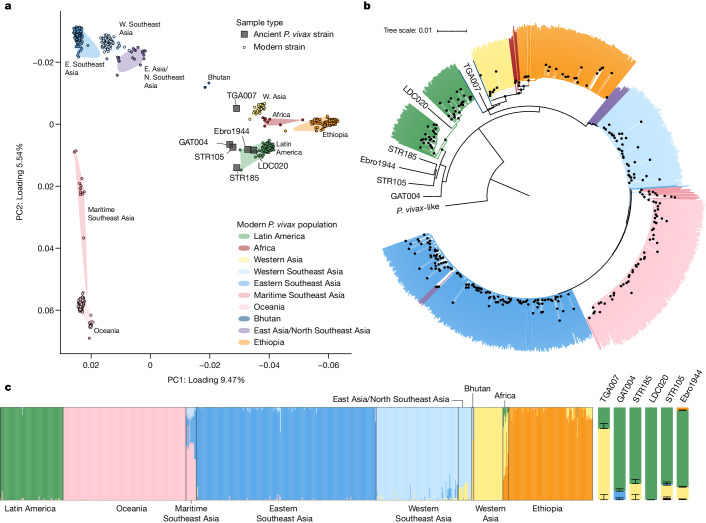
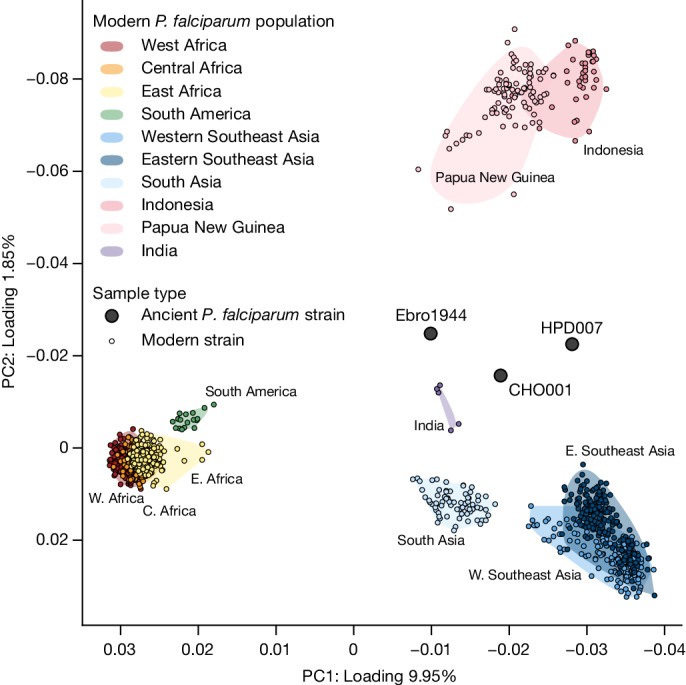
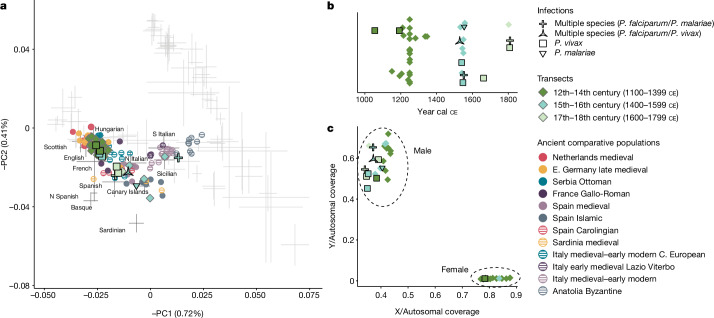
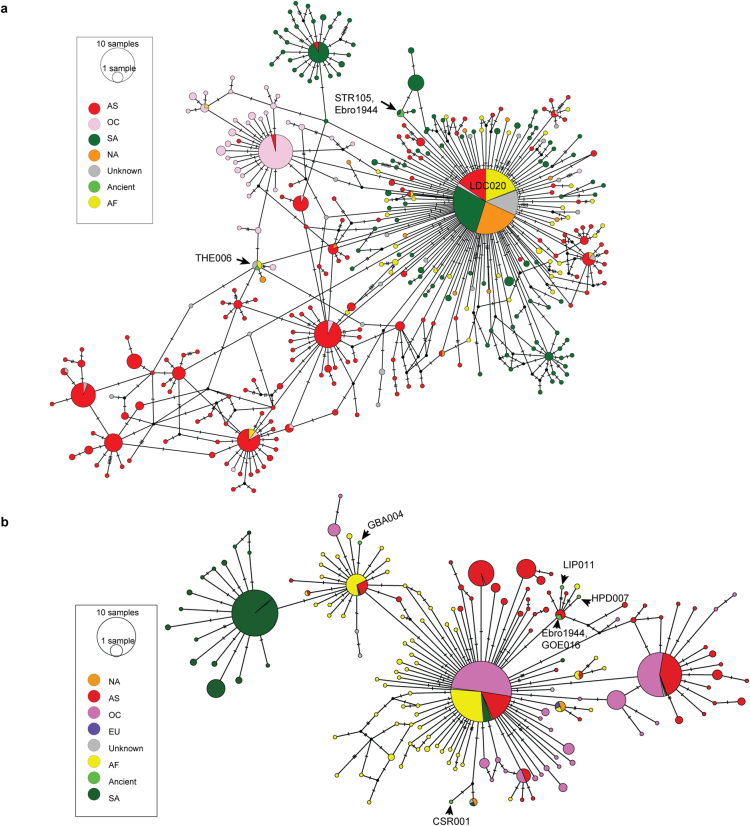
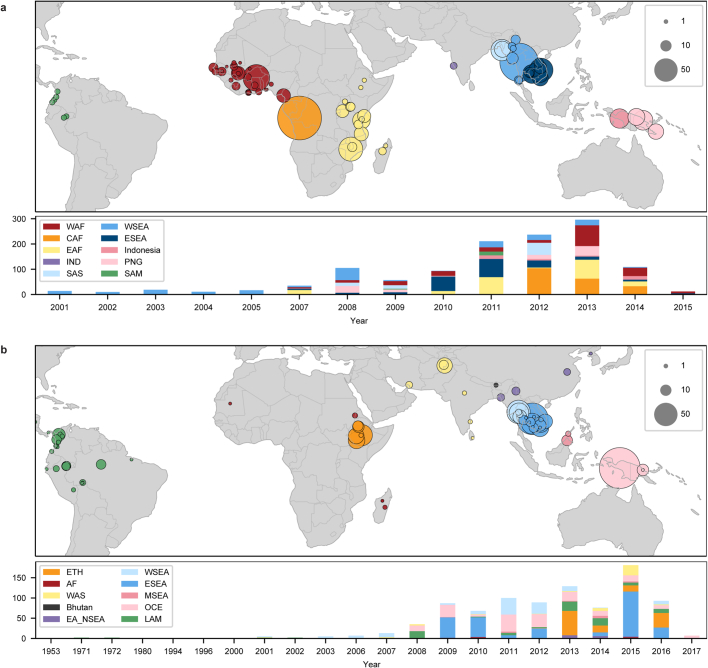
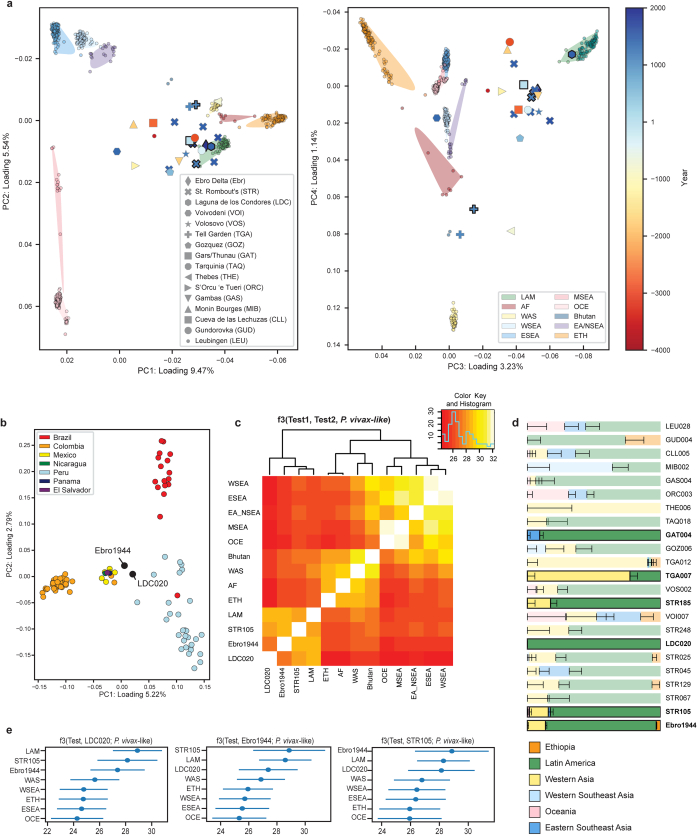
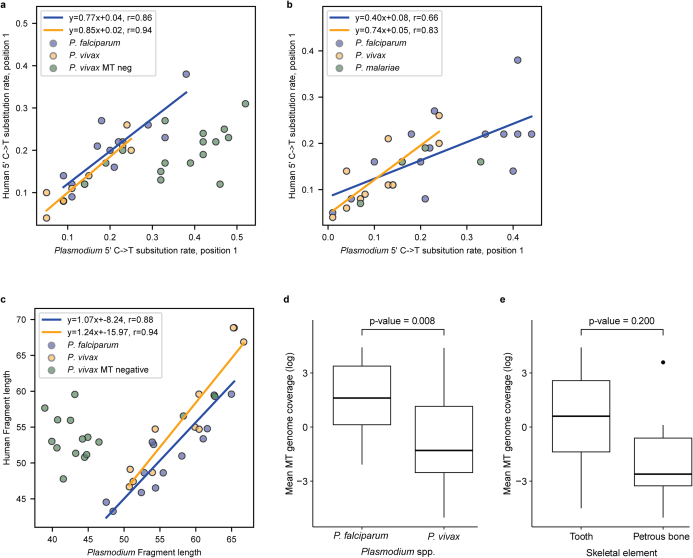
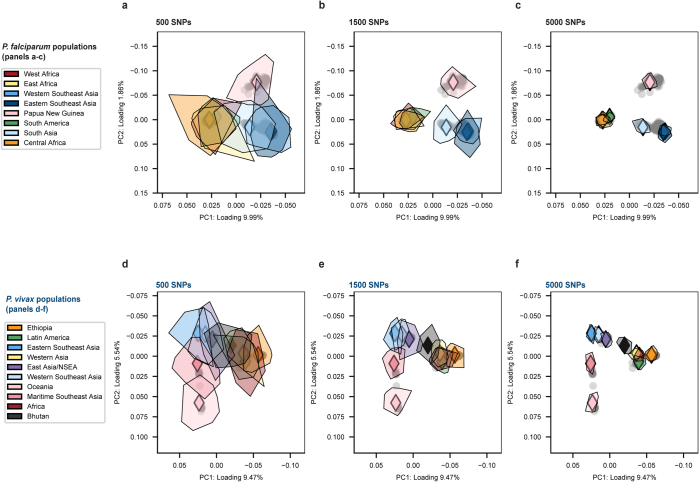
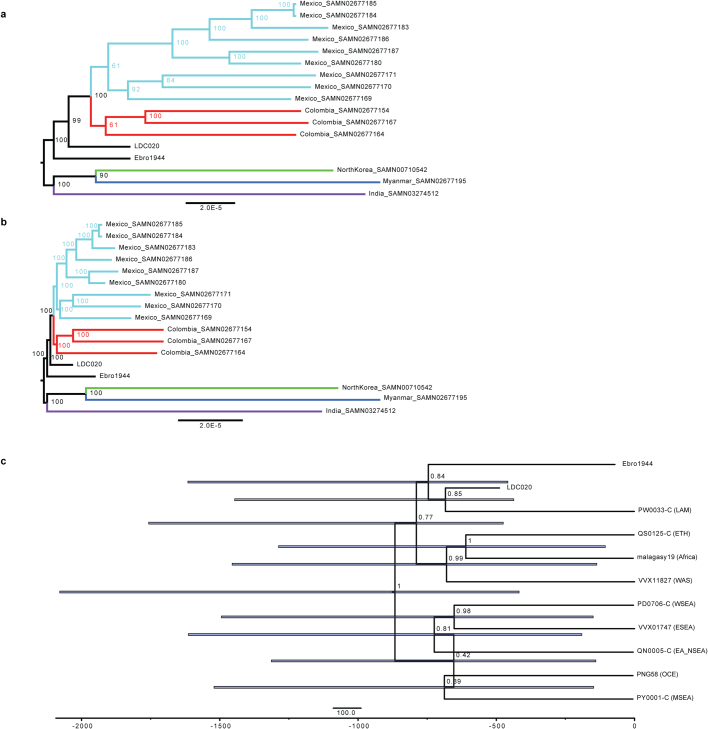
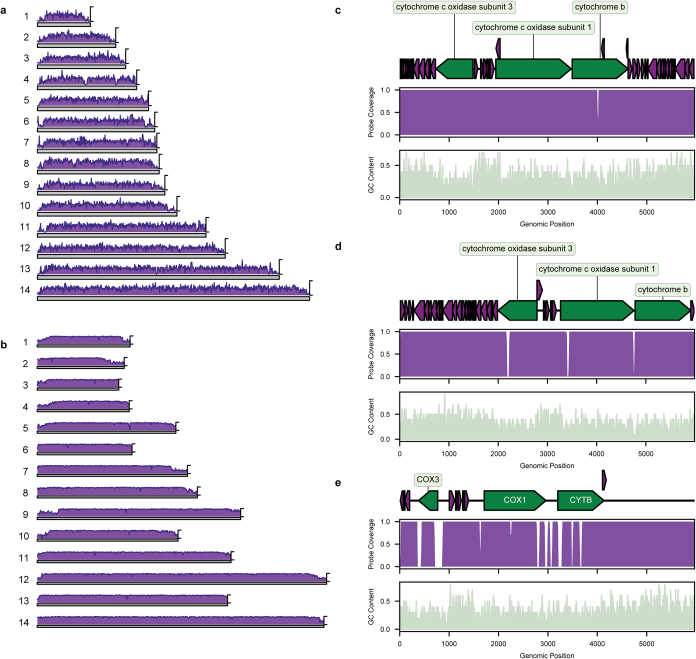
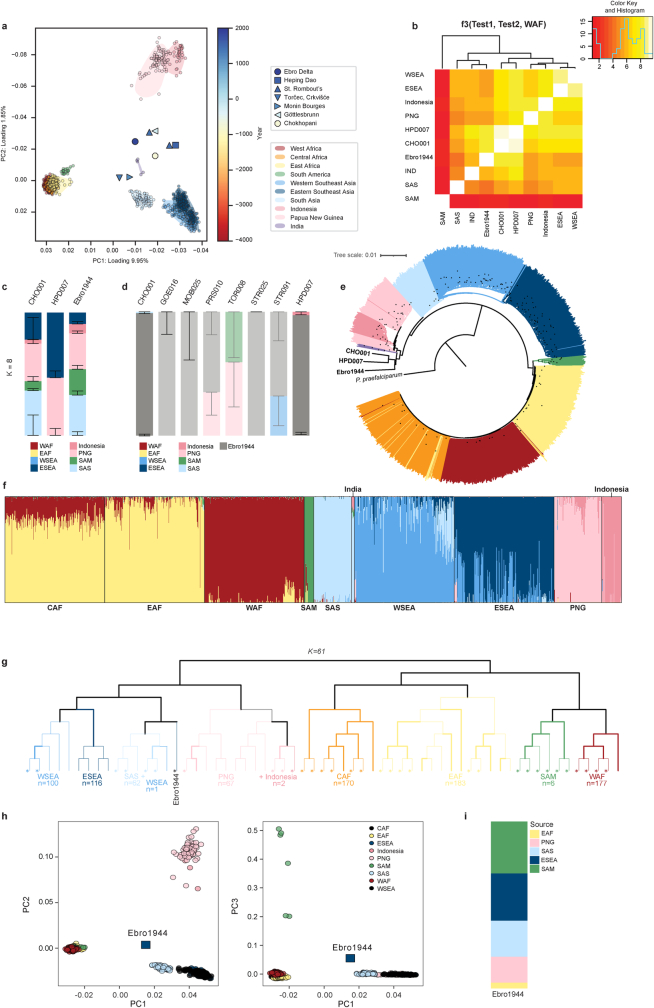
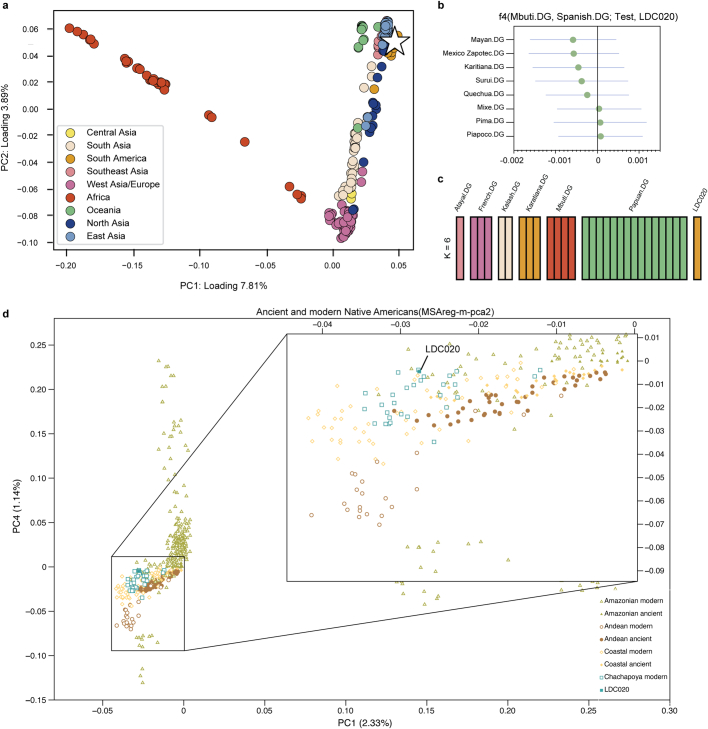
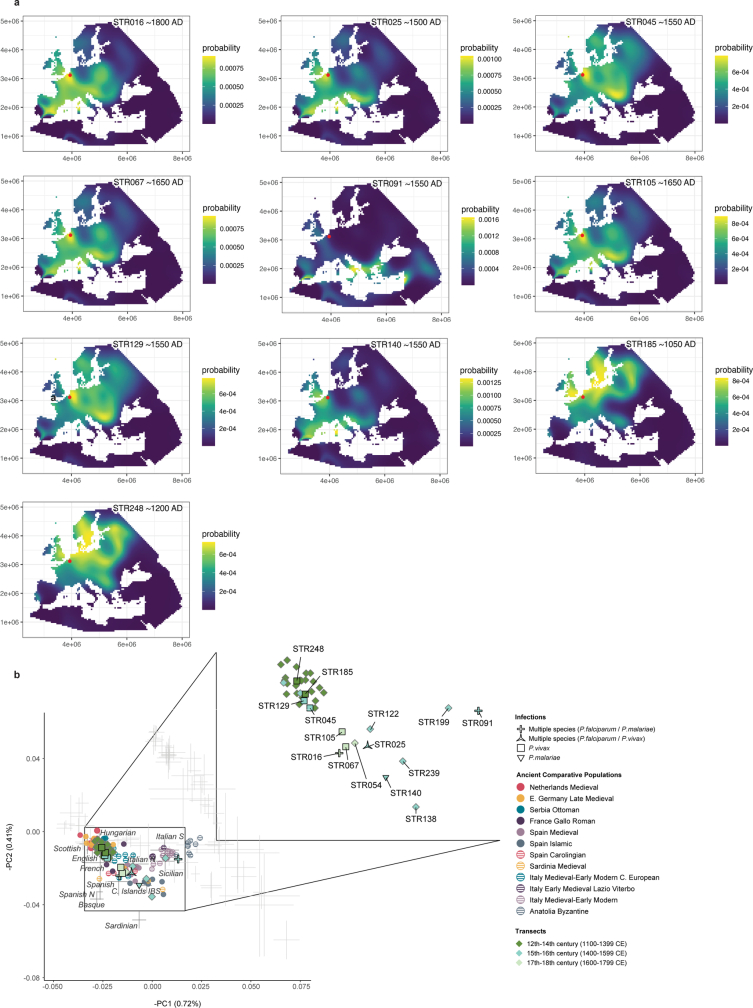
Malaria-causing protozoa of the genus Plasmodium have exerted one of the strongest selective pressures on the human genome, and resistance alleles provide biomolecular footprints that outline the historical reach of these species1. Nevertheless, debate persists over when and how malaria parasites emerged as human pathogens and spread around the globe1,2. To address these questions, we generated high-coverage ancient mitochondrial and nuclear genome-wide data from P. falciparum, P. vivax and P. malariae from 16 countries spanning around 5,500 years of human history. We identified P. vivax and P. falciparum across geographically disparate regions of Eurasia from as early as the fourth and first millennia BCE, respectively; for P. vivax, this evidence pre-dates textual references by several millennia3. Genomic analysis supports distinct disease histories for P. falciparum and P. vivax in the Americas: similarities between now-eliminated European and peri-contact South American strains indicate that European colonizers were the source of American P. vivax, whereas the trans-Atlantic slave trade probably introduced P. falciparum into the Americas. Our data underscore the role of cross-cultural contacts in the dissemination of malaria, laying the biomolecular foundation for future palaeo-epidemiological research into the impact of Plasmodium parasites on human history. Finally, our unexpected discovery of P. falciparum in the high-altitude Himalayas provides a rare case study in which individual mobility can be inferred from infection status, adding to our knowledge of cross-cultural connectivity in the region nearly three millennia ago.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Epidemiologic aspects of the malaria transmission cycle in an area of very low incidence in Brazil.Malar J. 2007 Mar 19;6:33. doi: 10.1186/1475-2875-6-33. Malar J. 2007. PMID: 17371598 Free PMC article.
-
Molecular evidence of high rates of asymptomatic P. vivax infection and very low P. falciparum malaria in Botswana.BMC Infect Dis. 2016 Sep 29;16(1):520. doi: 10.1186/s12879-016-1857-8. BMC Infect Dis. 2016. PMID: 27682611 Free PMC article.
-
Plasmodium malariae and Plasmodium ovale infections in the China-Myanmar border area.Malar J. 2016 Nov 15;15(1):557. doi: 10.1186/s12936-016-1605-y. Malar J. 2016. PMID: 27846879 Free PMC article.
-
Out of Africa: origins and evolution of the human malaria parasites Plasmodium falciparum and Plasmodium vivax.Int J Parasitol. 2017 Feb;47(2-3):87-97. doi: 10.1016/j.ijpara.2016.05.008. Epub 2016 Jul 2. Int J Parasitol. 2017. PMID: 27381764 Free PMC article. Review.
-
Genetic diversity and population history of Plasmodium falciparum and Plasmodium vivax.Parassitologia. 2006 Dec;48(4):561-6. Parassitologia. 2006. PMID: 17688177 Review. No abstract available.
Cited by
-
Intra-individual variability in ancient plasmodium DNA recovery highlights need for enhanced sampling.Sci Rep. 2025 Jan 4;15(1):757. doi: 10.1038/s41598-024-85038-z. Sci Rep. 2025. PMID: 39755798 Free PMC article.
-
Malaria: past, present, and future.Signal Transduct Target Ther. 2025 Jun 17;10(1):188. doi: 10.1038/s41392-025-02246-3. Signal Transduct Target Ther. 2025. PMID: 40523953 Free PMC article. Review.
-
Global Fight against Malaria: Goals and Achievements 1900-2022.J Clin Med. 2024 Sep 24;13(19):5680. doi: 10.3390/jcm13195680. J Clin Med. 2024. PMID: 39407740 Free PMC article. Review.
-
Current and future temperature suitability for autochthonous transmission of malaria in Canada.Int J Health Geogr. 2025 Aug 6;24(1):21. doi: 10.1186/s12942-025-00407-9. Int J Health Geogr. 2025. PMID: 40770641 Free PMC article.
-
Genomic exploration of the journey of Plasmodium vivax in Latin America.PLoS Pathog. 2025 Jan 13;21(1):e1012811. doi: 10.1371/journal.ppat.1012811. eCollection 2025 Jan. PLoS Pathog. 2025. PMID: 39804931 Free PMC article.
References
-
- Ashley EA, Pyae Phyo A, Woodrow CJ. Malaria. Lancet. 2018;391:1608–1621. - PubMed
-
- World Health Organization. World Malaria Report 2021 (World Health Organization, 2021).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
