

Clinical, radiological and histopathological features of patients with familial pulmonary fibrosis
- PMID: 38867203
- PMCID: PMC11170837
- DOI: 10.1186/s12931-024-02864-5
Clinical, radiological and histopathological features of patients with familial pulmonary fibrosis
Abstract
Background: In familial pulmonary fibrosis (FPF) at least two biological relatives are affected. Patients with FPF have diverse clinical features.
Research question: We aimed to characterize demographic and clinical features, re-evaluate high-resolution computed tomography (HRCT) scans and histopathology of surgical lung biopsies, assess survival and investigate the suitability of risk prediction models for FPF patients.
Study design: A retrospective cohort study.
Methods: FPF data (n = 68) were collected from the medical records of Oulu University Hospital (OUH) and Oulaskangas District Hospital between 1 Jan 2000 and 11 Jan 2023. The inclusion criterion was pulmonary fibrosis (PF) (ICD 10-code J84.X) and at least one self-reported relative with PF. Clinical information was gathered from hospital medical records. HRCT scans and histology were re-evaluated.
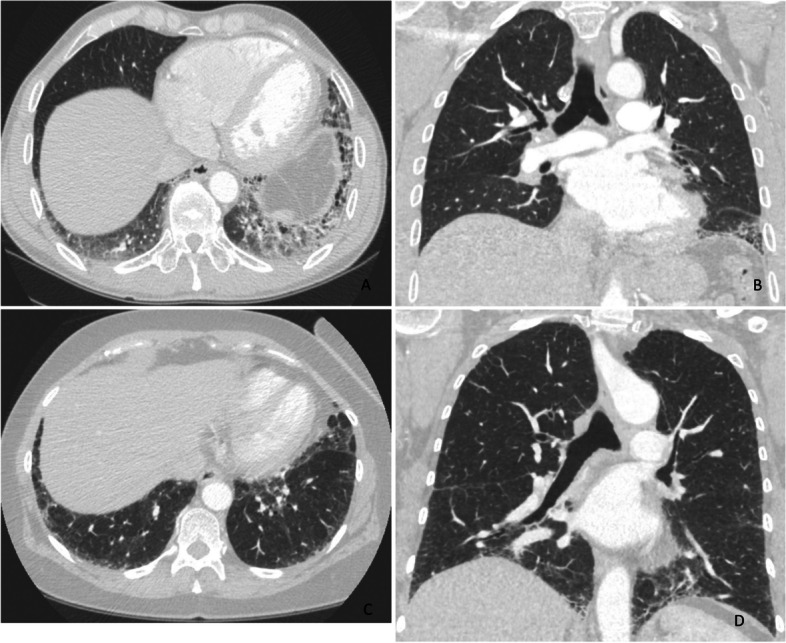
Results: Thirty-seven (54.4%) of the patients were men, and 31 (45.6%) were women. The mean ages of the women and men were 68.6 and 61.7 years, respectively (p = 0.003). Thirty-seven (54.4%) patients were nonsmokers. The most common radiological patterns were usual interstitial pneumonia (UIP) (51/75.0%), unclassifiable (8/11.8%) and nonspecific interstitial pneumonia (NSIP) (3/4.4%). Pleuroparenchymal fibroelastosis (PPFE) was observed as a single or combined pattern in 13.2% of the patients. According to the 2022 guidelines for idiopathic pulmonary fibrosis (IPF), the patients were categorized as UIP (31/45.6%), probable UIP (20/29.4%), indeterminate for UIP (7/10.3%) or alternative diagnosis (10/14.7%). The histopathological patterns were UIP (7/41.2%), probable UIP (1/5.9%), indeterminate for UIP (8/47.2%) and alternative diagnosis (1/5.9%). Rare genetic variants were found in 9 patients; these included telomerase reverse transcriptase (TERT, n = 6), telomerase RNA component (TERC, n = 2) and regulator of telomere elongation helicase 1 (RTEL1, n = 1). Half of the patients died (n = 29) or underwent lung transplantation (n = 5), with a median survival of 39.9 months. The risk prediction models composite physiology index (CPI), hazard ratio (HR) 1.07 (95.0% CI 1.04-1.10), and gender-age-physiology index (GAP) stage I predicted survival statistically significantly (p<0.001) compared to combined stages II and III.
Conclusions: This study confirmed the results of earlier studies showing that FPF patients' radiological and histopathological patterns are diverse. Moreover, radiological and histological features revealed unusual patterns and their combinations.
Keywords: Comorbidity; Familial pulmonary fibrosis; Histopathology; Interstitial lung disease; Radiology; Risk prediction model; Survival.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
