

How much metagenome data is needed for protein structure prediction: The advantages of targeted approach from the ecological and evolutionary perspectives
- PMID: 38867727
- PMCID: PMC10989767
- DOI: 10.1002/imt2.9
How much metagenome data is needed for protein structure prediction: The advantages of targeted approach from the ecological and evolutionary perspectives
Abstract
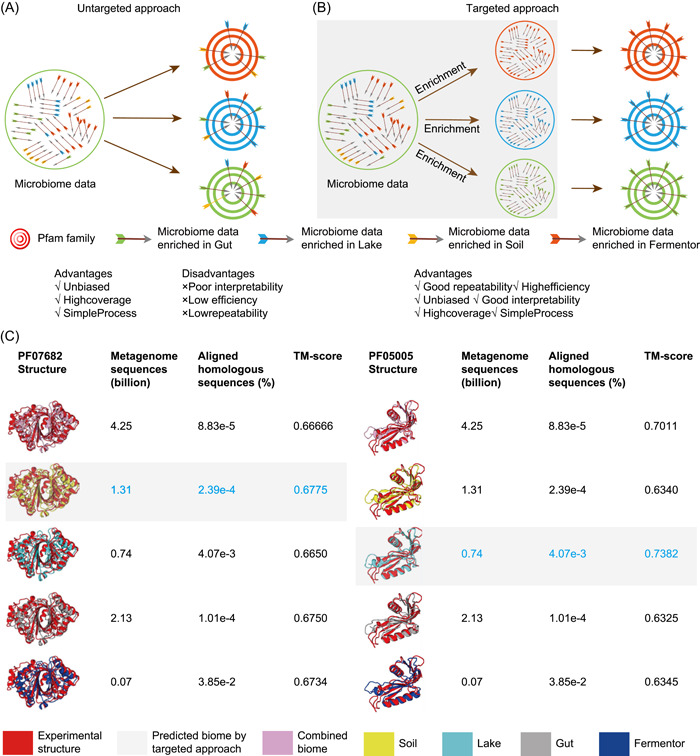
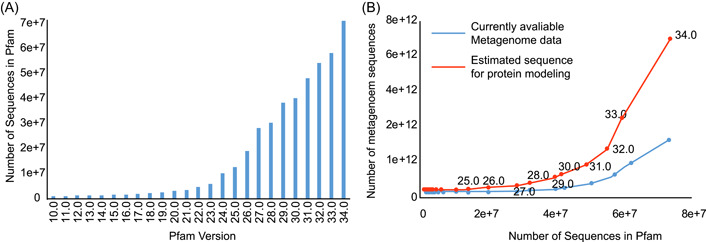
It has been proven that three-dimensional protein structures could be modeled by supplementing homologous sequences with metagenome sequences. Even though a large volume of metagenome data is utilized for such purposes, a significant proportion of proteins remain unsolved. In this review, we focus on identifying ecological and evolutionary patterns in metagenome data, decoding the complicated relationships of these patterns with protein structures, and investigating how these patterns can be effectively used to improve protein structure prediction. First, we proposed the metagenome utilization efficiency and marginal effect model to quantify the divergent distribution of homologous sequences for the protein family. Second, we proposed that the targeted approach effectively identifies homologous sequences from specified biomes compared with the untargeted approach's blind search. Finally, we determined the lower bound for metagenome data required for predicting all the protein structures in the Pfam database and showed that the present metagenome data is insufficient for this purpose. In summary, we discovered ecological and evolutionary patterns in the metagenome data that may be used to predict protein structures effectively. The targeted approach is promising in terms of effectively extracting homologous sequences and predicting protein structures using these patterns.
Keywords: ecology; evolution; metagenome data; protein 3D structure modeling; targeted approach.
© 2022 The Authors. iMeta published by John Wiley & Sons Australia, Ltd on behalf of iMeta Science.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Decoding the link of microbiome niches with homologous sequences enables accurately targeted protein structure prediction.Proc Natl Acad Sci U S A. 2021 Dec 7;118(49):e2110828118. doi: 10.1073/pnas.2110828118. Proc Natl Acad Sci U S A. 2021. PMID: 34873061 Free PMC article.
-
MetCap: a bioinformatics probe design pipeline for large-scale targeted metagenomics.BMC Bioinformatics. 2015 Feb 28;16(1):65. doi: 10.1186/s12859-015-0501-8. BMC Bioinformatics. 2015. PMID: 25880302 Free PMC article.
-
SUPFAM--a database of potential protein superfamily relationships derived by comparing sequence-based and structure-based families: implications for structural genomics and function annotation in genomes.Nucleic Acids Res. 2002 Jan 1;30(1):289-93. doi: 10.1093/nar/30.1.289. Nucleic Acids Res. 2002. PMID: 11752317 Free PMC article.
-
Integrating pan-genome with metagenome for microbial community profiling.Comput Struct Biotechnol J. 2021 Mar 7;19:1458-1466. doi: 10.1016/j.csbj.2021.02.021. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 33841754 Free PMC article. Review.
-
Targeted metagenomics: a high-resolution metagenomics approach for specific gene clusters in complex microbial communities.Environ Microbiol. 2012 Jan;14(1):13-22. doi: 10.1111/j.1462-2920.2011.02438.x. Epub 2011 Mar 1. Environ Microbiol. 2012. PMID: 21366818 Review.
Cited by
-
iMeta: Integrated meta-omics for biology and environments.Imeta. 2022 Mar 28;1(1):e15. doi: 10.1002/imt2.15. eCollection 2022 Mar. Imeta. 2022. PMID: 38867730 Free PMC article.
-
Leveraging computer-aided design and artificial intelligence to develop a next-generation multi-epitope tuberculosis vaccine candidate.Infect Med (Beijing). 2024 Nov 9;3(4):100148. doi: 10.1016/j.imj.2024.100148. eCollection 2024 Dec. Infect Med (Beijing). 2024. PMID: 39687693 Free PMC article.
-
MicroEXPERT: Microbiome profiling platform with cross-study metagenome-wide association analysis functionality.Imeta. 2023 Aug 17;2(4):e131. doi: 10.1002/imt2.131. eCollection 2023 Nov. Imeta. 2023. PMID: 38868224 Free PMC article.
References
-
- Zhang, Chengxin , Zheng Wei, Freddolino Peter L., and Zhang Yang. 2018. “MetaGO: Predicting Gene Ontology of Non‐Homologous Proteins Through Low‐Resolution Protein Structure Prediction and Protein‐Protein Network Mapping.” Journal of Molecular Biology 430: 2256–65. 10.1016/j.jmb.2018.03.004 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources