

OpenSTED: open-source dynamic intensity minimum system for stimulated emission depletion microscopy
- PMID: 38867758
- PMCID: PMC11167952
- DOI: 10.1117/1.NPh.11.3.034311
OpenSTED: open-source dynamic intensity minimum system for stimulated emission depletion microscopy
Abstract
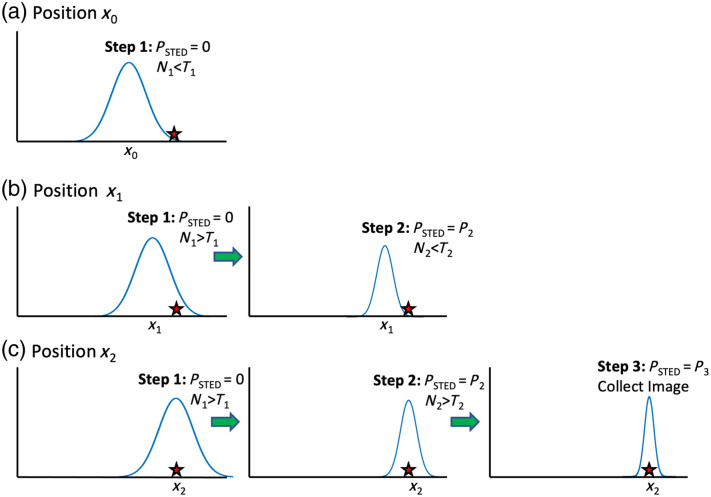
Significance: Stimulated emission depletion (STED) is a powerful super-resolution microscopy technique that can be used for imaging live cells. However, the high STED laser powers can cause significant photobleaching and sample damage in sensitive biological samples. The dynamic intensity minimum (DyMIN) technique turns on the STED laser only in regions of the sample where there is fluorescence signal, thus saving significant sample photobleaching. The reduction in photobleaching allows higher resolution images to be obtained and longer time-lapse imaging of live samples. A stand-alone module to perform DyMIN is not available commercially.
Aim: In this work, we developed an open-source design to implement three-step DyMIN on a STED microscope and demonstrated reduced photobleaching for timelapse imaging of beads, cells, and tissue.
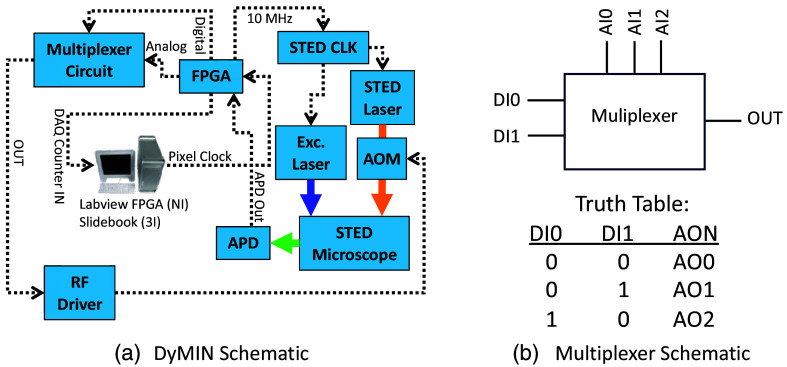
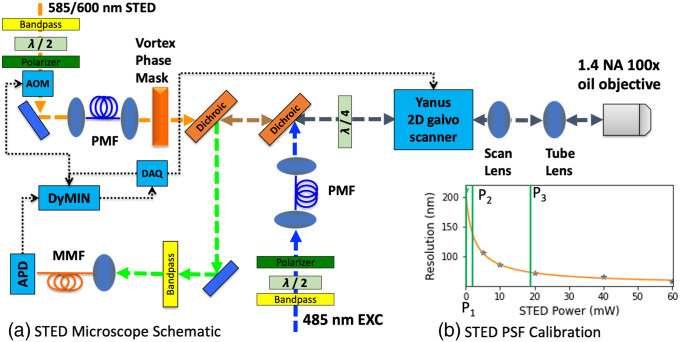
Approach: The DyMIN system uses a fast multiplexer circuit and inexpensive field-programmable gate array controlled by Labview software that operates as a stand-alone module for a STED microscope. All software and circuit diagrams are freely available.
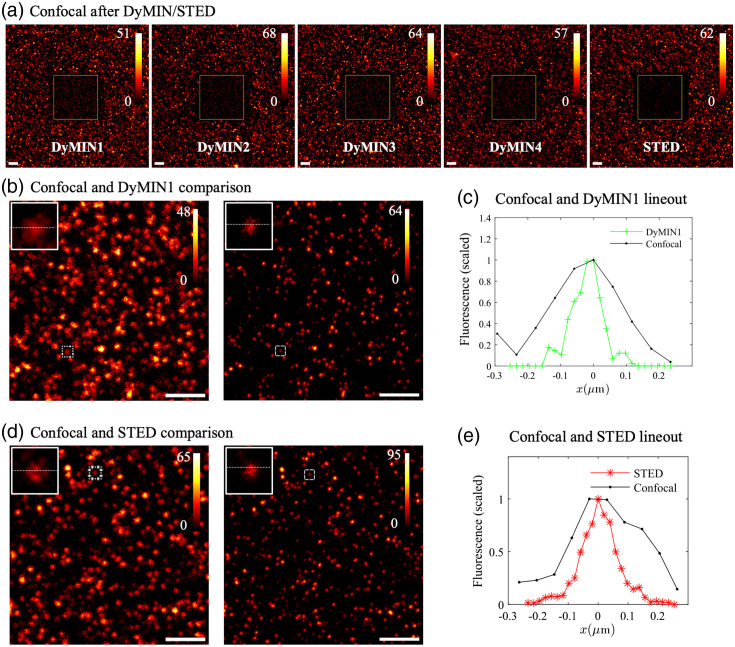
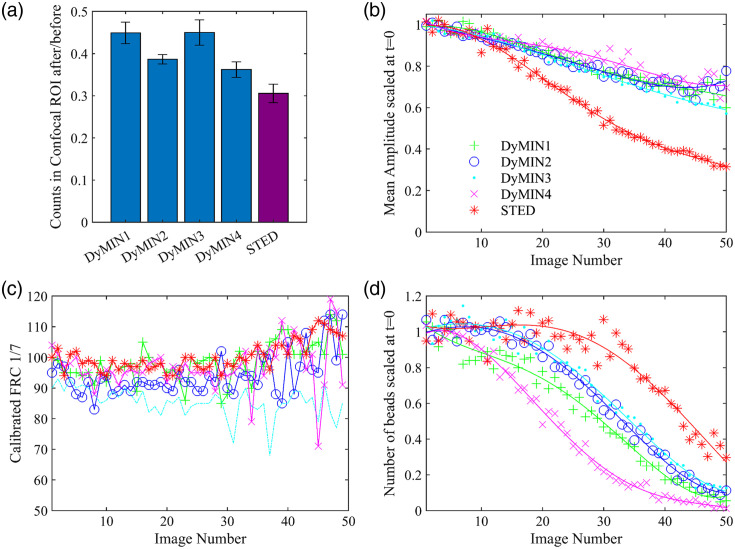
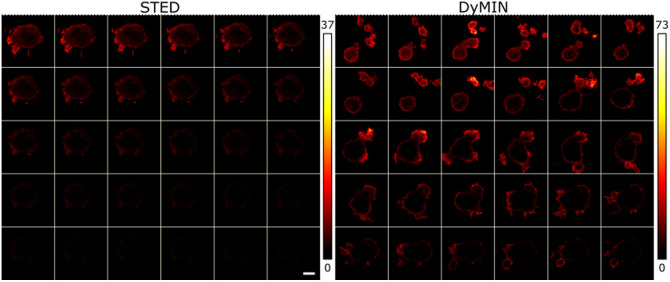
Results: We compared time-lapse images of bead samples using our custom DyMIN system to conventional STED and recorded a higher signal when using DyMIN after a 50-image sequence. We further demonstrated the DyMIN system for time-lapse STED imaging of live cells and brain tissue slices.
Conclusions: Our open-source DyMIN system is an inexpensive add-on to a conventional STED microscope that can reduce photobleaching. The system can significantly improve signal to noise for dynamic time-lapse STED imaging of live samples.
Keywords: dynamic minimum; fluorescence; microscopy; stimulated emission depletion; super-resolution.
© 2024 The Authors.
Figures

Similar articles
-
Adaptive-illumination STED nanoscopy.Proc Natl Acad Sci U S A. 2017 Sep 12;114(37):9797-9802. doi: 10.1073/pnas.1708304114. Epub 2017 Aug 28. Proc Natl Acad Sci U S A. 2017. PMID: 28847959 Free PMC article.
-
Photobleaching reduction in modulated super-resolution microscopy.Microscopy (Oxf). 2021 Jun 6;70(3):278-288. doi: 10.1093/jmicro/dfaa062. Microscopy (Oxf). 2021. PMID: 33064828
-
Low-Power Two-Color Stimulated Emission Depletion Microscopy for Live Cell Imaging.Biosensors (Basel). 2021 Sep 10;11(9):330. doi: 10.3390/bios11090330. Biosensors (Basel). 2021. PMID: 34562919 Free PMC article.
-
Strategies to maximize performance in STimulated Emission Depletion (STED) nanoscopy of biological specimens.Methods. 2020 Mar 1;174:27-41. doi: 10.1016/j.ymeth.2019.07.019. Epub 2019 Jul 22. Methods. 2020. PMID: 31344404 Free PMC article. Review.
-
Stochastic optical reconstruction microscopy (STORM) in comparison with stimulated emission depletion (STED) and other imaging methods.J Neurochem. 2015 Nov;135(4):643-58. doi: 10.1111/jnc.13257. Epub 2015 Sep 14. J Neurochem. 2015. PMID: 26222552 Review.
Cited by
-
Special Section Guest Editorial: Open-source neurophotonic tools for neuroscience.Neurophotonics. 2024 Jul;11(3):034301. doi: 10.1117/1.NPh.11.3.034301. Epub 2024 Sep 30. Neurophotonics. 2024. PMID: 39350913 Free PMC article.
