

Speckle-enabled in vivo demixing of neural activity in the mouse brain
- PMID: 38867774
- PMCID: PMC11166431
- DOI: 10.1364/BOE.524521
Speckle-enabled in vivo demixing of neural activity in the mouse brain
Abstract
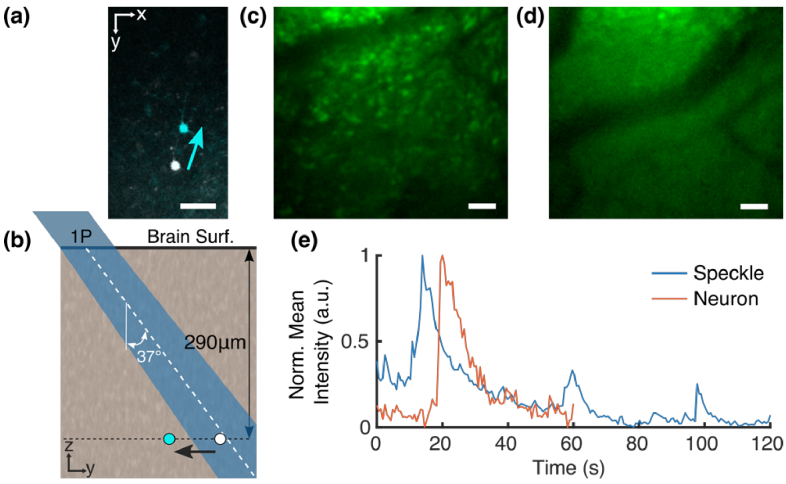
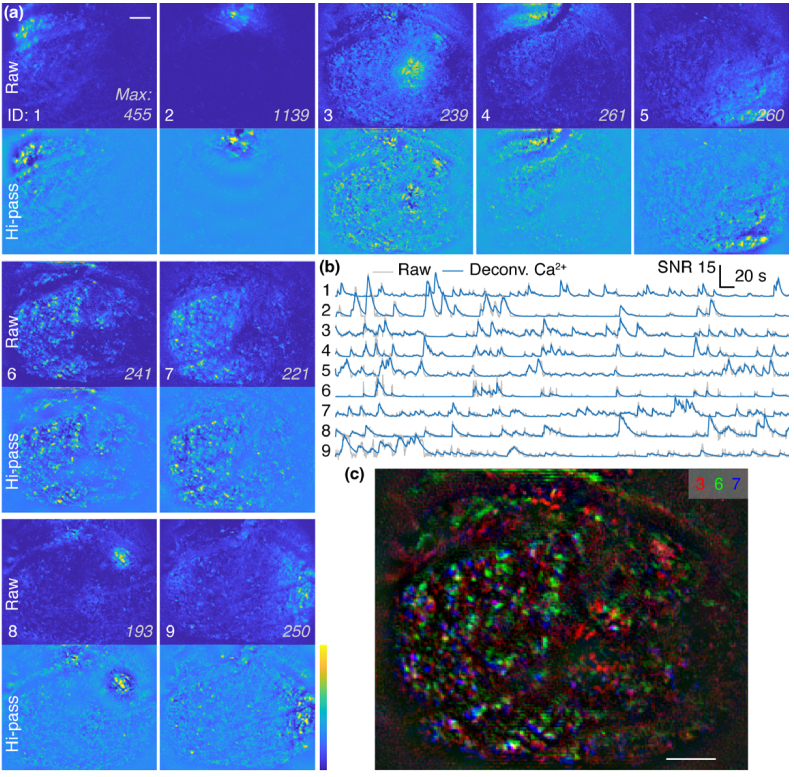
Functional imaging of neuronal activity in awake animals, using a combination of fluorescent reporters of neuronal activity and various types of microscopy modalities, has become an indispensable tool in neuroscience. While various imaging modalities based on one-photon (1P) excitation and parallel (camera-based) acquisition have been successfully used for imaging more transparent samples, when imaging mammalian brain tissue, due to their scattering properties, two-photon (2P) microscopy systems are necessary. In 2P microscopy, the longer excitation wavelengths reduce the amount of scattering while the diffraction-limited 3D localization of excitation largely eliminates out-of-focus fluorescence. However, this comes at the cost of time-consuming serial scanning of the excitation spot and more complex and expensive instrumentation. Thus, functional 1P imaging modalities that can be used beyond the most transparent specimen are highly desirable. Here, we transform light scattering from an obstacle into a tool. We use speckles with their unique patterns and contrast, formed when fluorescence from individual neurons propagates through rodent cortical tissue, to encode neuronal activity. Spatiotemporal demixing of these patterns then enables functional recording of neuronal activity from a group of discriminable sources. For the first time, we provide an experimental, in vivo characterization of speckle generation, speckle imaging and speckle-assisted demixing of neuronal activity signals in the scattering mammalian brain tissue. We found that despite an initial fast speckle decorrelation, substantial correlation was maintained over minute-long timescales that contributed to our ability to demix temporal activity traces in the mouse brain in vivo. Informed by in vivo quantifications of speckle patterns from single and multiple neurons excited using 2P scanning excitation, we recorded and demixed activity from several sources excited using 1P oblique illumination. In our proof-of-principle experiments, we demonstrate in vivo speckle-assisted demixing of functional signals from groups of sources in a depth range of 220-320 µm in mouse cortex, limited by available speckle contrast. Our results serve as a basis for designing an in vivo functional speckle imaging modality and for maximizing the key resource in any such modality, the speckle contrast. We anticipate that our results will provide critical quantitative guidance to the community for designing techniques that overcome light scattering as a fundamental limitation in bioimaging.
© 2024 Optica Publishing Group.
Conflict of interest statement
The authors declare that there are no conflicts of interest related to this article.
Figures







References
Grants and funding
LinkOut - more resources
Full Text Sources