

Myricanol prevents aging-related sarcopenia by rescuing mitochondrial dysfunction via targeting peroxiredoxin 5
- PMID: 38868327
- PMCID: PMC11167181
- DOI: 10.1002/mco2.566
Myricanol prevents aging-related sarcopenia by rescuing mitochondrial dysfunction via targeting peroxiredoxin 5
Abstract
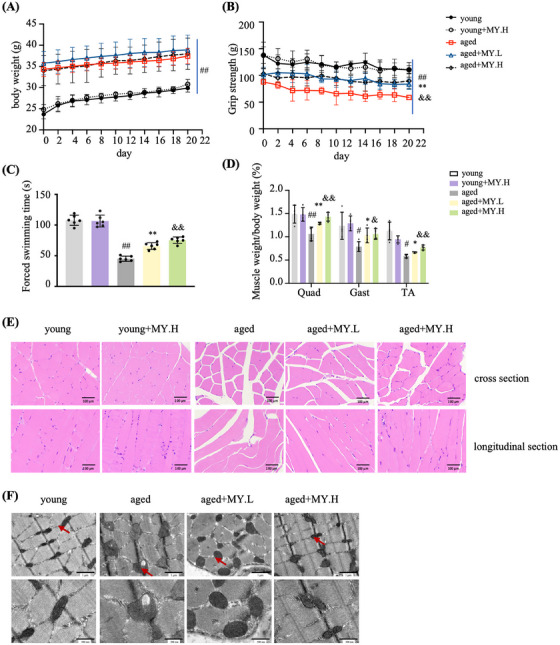
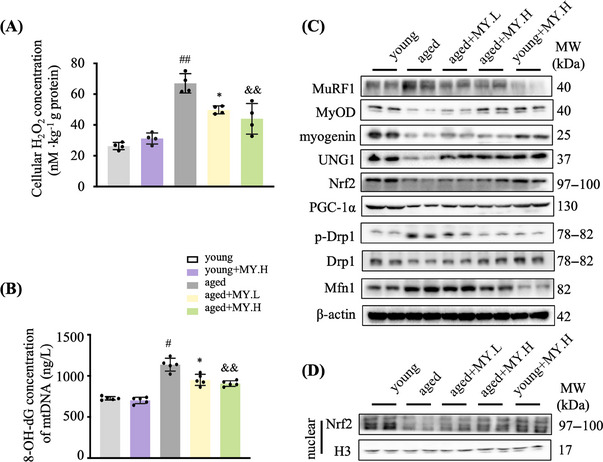
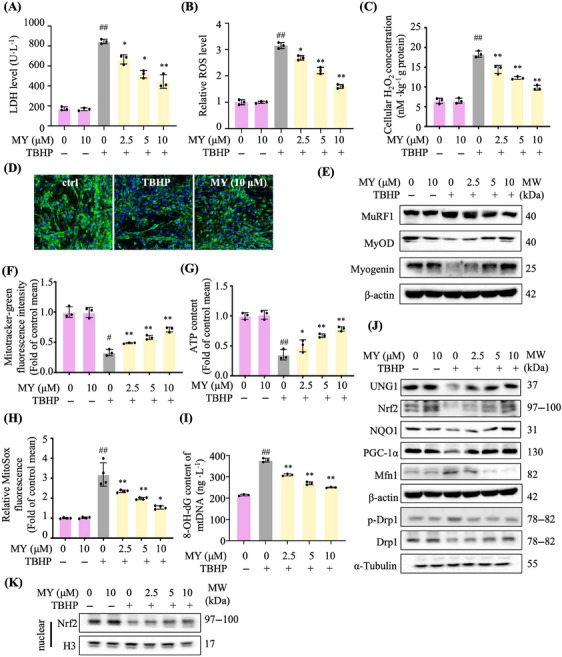
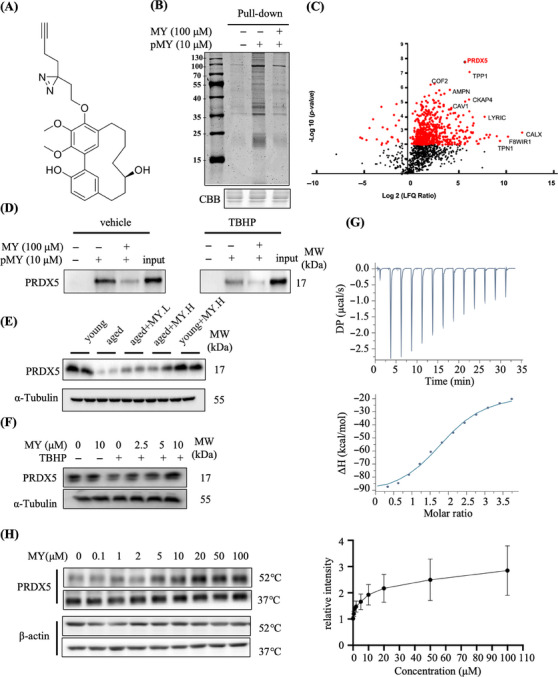
Aging is a process that represents the accumulation of changes in organism overtime. In biological level, accumulations of molecular and cellular damage in aging lead to an increasing risk of diseases like sarcopenia. Sarcopenia reduces mobility, leads to fall-related injuries, and diminishes life quality. Thus, it is meaningful to find out novel therapeutic strategies for sarcopenia intervention that may help the elderly maintain their functional ability. Oxidative damage-induced dysfunctional mitochondria are considered as a culprit of muscle wasting during aging. Herein, we aimed to demonstrate whether myricanol (MY) protects aged mice against muscle wasting through alleviating oxidative damage in mitochondria and identify the direct protein target and its underlying mechanism. We discovered that MY protects aged mice against the loss of muscle mass and strength through scavenging reactive oxygen species accumulation to rebuild the redox homeostasis. Taking advantage of biophysical assays, peroxiredoxin 5 was discovered and validated as the direct target of MY. Through activating peroxiredoxin 5, MY reduced reactive oxygen species accumulation and damaged mitochondrial DNA in C2C12 myotubes. Our findings provide an insight for therapy against sarcopenia through alleviating oxidative damage-induced dysfunctional mitochondria by targeting peroxiredoxin 5, which may contribute an insight for healthy aging.
Keywords: healthy aging; mitochondria; myricanol; peroxiredoxin 5; redox homeostasis; sarcopenia.
© 2024 The Author(s). MedComm published by Sichuan International Medical Exchange & Promotion Association (SCIMEA) and John Wiley & Sons Australia, Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
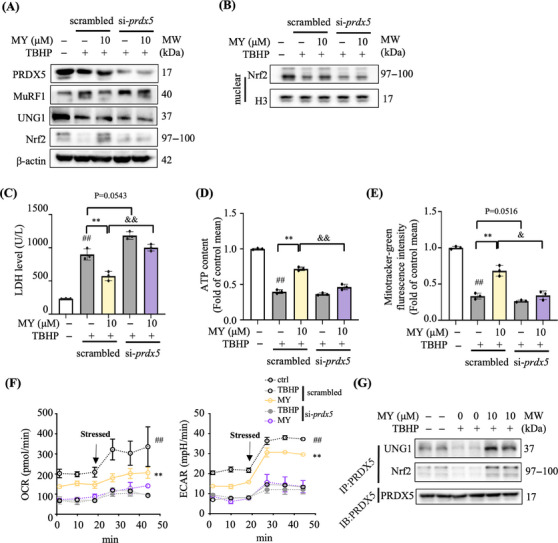
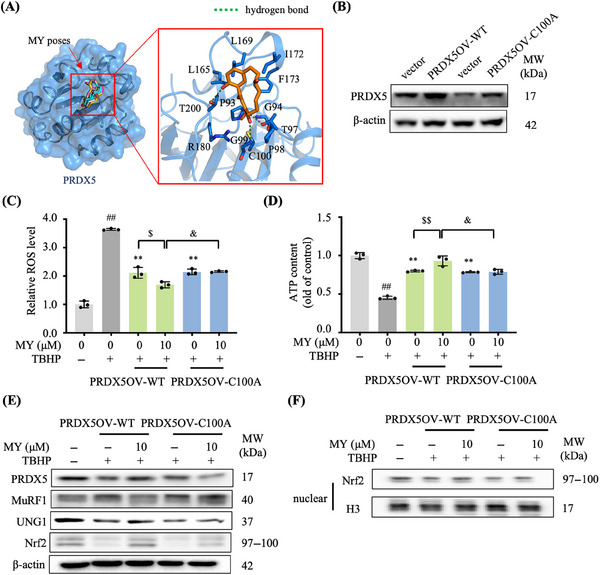
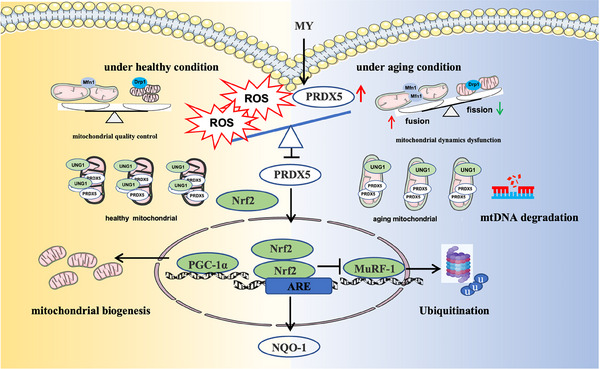
Similar articles
-
Myricanol rescues dexamethasone-induced muscle dysfunction via a sirtuin 1-dependent mechanism.J Cachexia Sarcopenia Muscle. 2019 Apr;10(2):429-444. doi: 10.1002/jcsm.12393. Epub 2019 Feb 21. J Cachexia Sarcopenia Muscle. 2019. PMID: 30793539 Free PMC article.
-
Scavenging mitochondrial hydrogen peroxide by peroxiredoxin 3 overexpression attenuates contractile dysfunction and muscle atrophy in a murine model of accelerated sarcopenia.Aging Cell. 2022 Mar;21(3):e13569. doi: 10.1111/acel.13569. Epub 2022 Feb 24. Aging Cell. 2022. PMID: 35199907 Free PMC article.
-
Polygonatum sibiricum polysaccharide ameliorates skeletal muscle aging via mitochondria-associated membrane-mediated calcium homeostasis regulation.Phytomedicine. 2024 Jul;129:155567. doi: 10.1016/j.phymed.2024.155567. Epub 2024 Mar 24. Phytomedicine. 2024. PMID: 38579644
-
From mitochondria to sarcopenia: Role of inflammaging and RAGE-ligand axis implication.Exp Gerontol. 2021 Apr;146:111247. doi: 10.1016/j.exger.2021.111247. Epub 2021 Jan 20. Exp Gerontol. 2021. PMID: 33484891 Review.
-
Mitochondrial Dynamics and Mitophagy in Skeletal Muscle Health and Aging.Int J Mol Sci. 2021 Jul 30;22(15):8179. doi: 10.3390/ijms22158179. Int J Mol Sci. 2021. PMID: 34360946 Free PMC article. Review.
Cited by
-
Exploring the role of traditional Chinese medicine in sarcopenia: mechanisms and therapeutic advances.Front Pharmacol. 2025 Jun 30;16:1541373. doi: 10.3389/fphar.2025.1541373. eCollection 2025. Front Pharmacol. 2025. PMID: 40661078 Free PMC article. Review.
-
The Organ-Joint Axes in Osteoarthritis: Significant Pathogenesis and Therapeutic Targets.Aging Dis. 2024 Nov 21;16(5):2999-3021. doi: 10.14336/AD.2024.1223. Aging Dis. 2024. PMID: 39656496 Free PMC article. Review.
-
Role of mitochondria in physiological activities, diseases, and therapy.Mol Biomed. 2025 Jun 19;6(1):42. doi: 10.1186/s43556-025-00284-5. Mol Biomed. 2025. PMID: 40536597 Free PMC article. Review.
References
-
- Cruz‐Jentoft AJ, Sarcopenia SA. Sarcopenia. Lancet. 2019;393(10191):2636‐2646. - PubMed
-
- Perez‐Lasierra JL, Casajus JA, González‐Agüero A, Moreno‐Franco B. Association of physical activity levels and prevalence of major degenerative diseases: evidence from the national health and nutrition examination survey (NHANES) 1999–2018. Exp Gerontol. 2022;158:111656. - PubMed
-
- Cleasby ME, Jamieson PM, Atherton PJ. Insulin resistance and sarcopenia: mechanistic links between common co‐morbidities. J Endocrinol. 2016;229(2):R67‐R81. - PubMed
-
- Sun D, Wang J, Toan S, et al. Molecular mechanisms of coronary microvascular endothelial dysfunction in diabetes mellitus: focus on mitochondrial quality surveillance. Angiogenesis. 2022;25(3):307‐329. - PubMed
LinkOut - more resources
Full Text Sources