

Alzheimer's disease and its treatment-yesterday, today, and tomorrow
- PMID: 38868666
- PMCID: PMC11167451
- DOI: 10.3389/fphar.2024.1399121
Alzheimer's disease and its treatment-yesterday, today, and tomorrow
Abstract
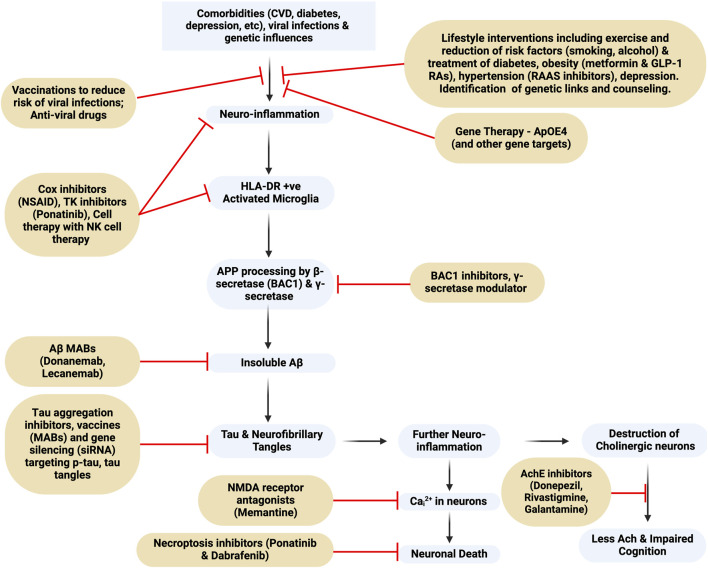
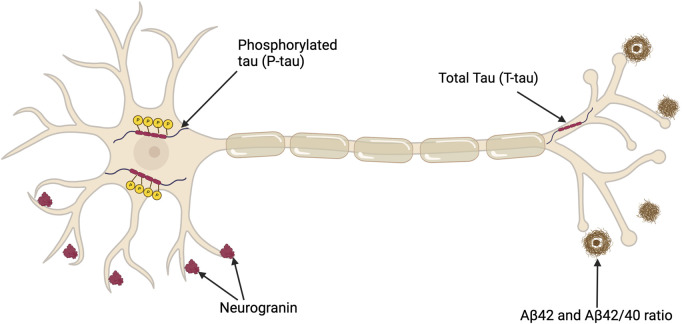
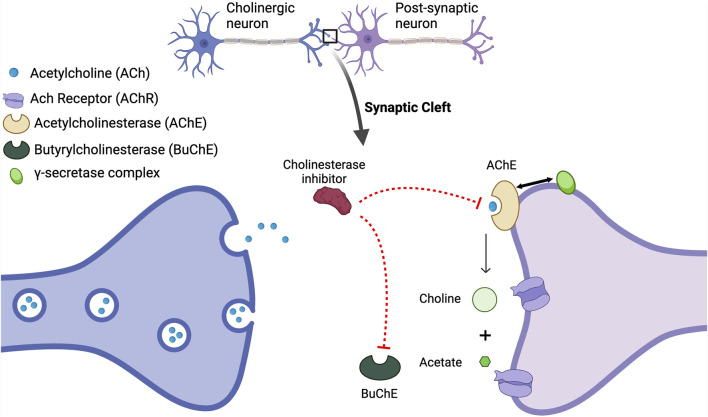
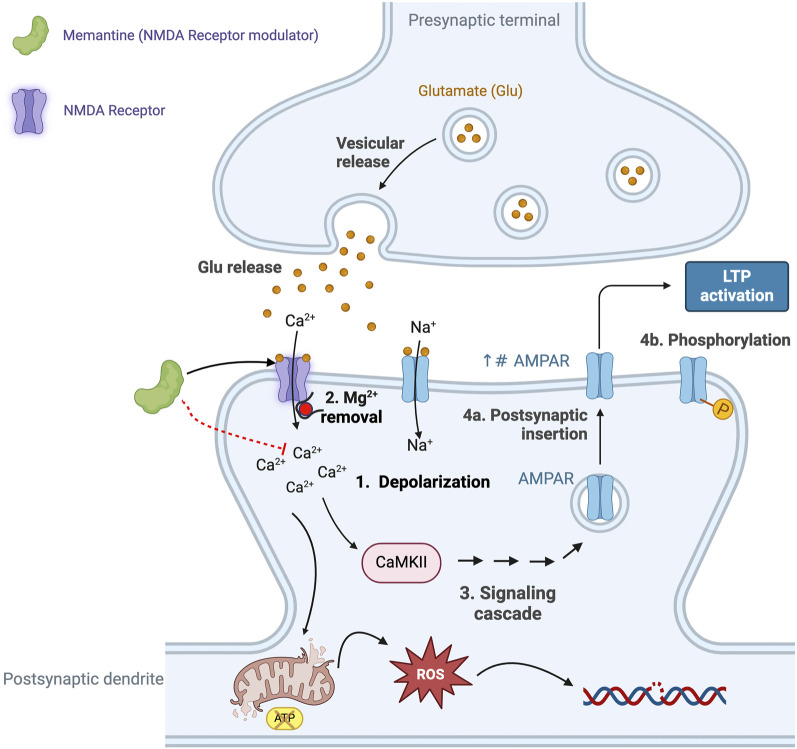
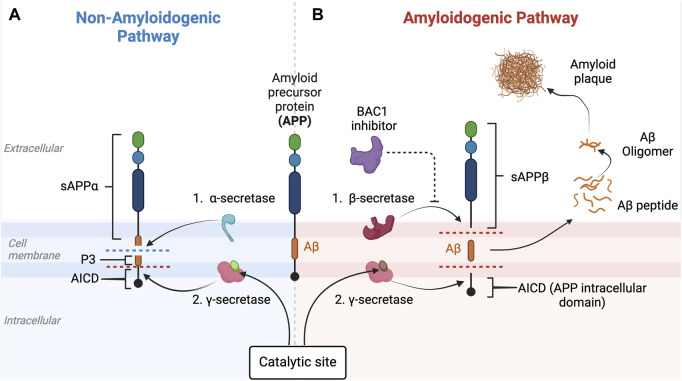
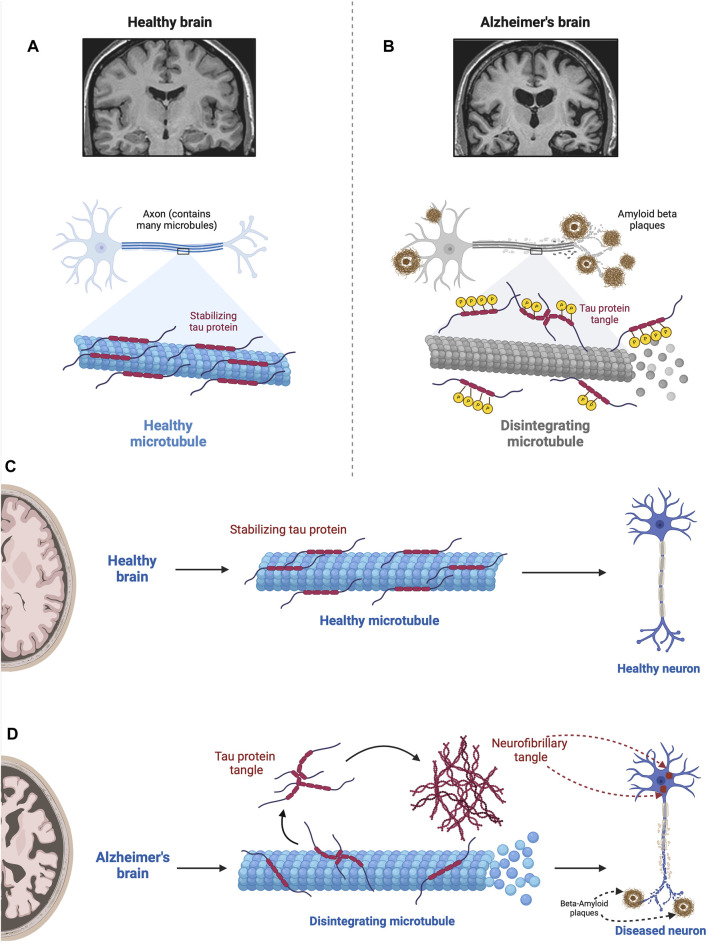
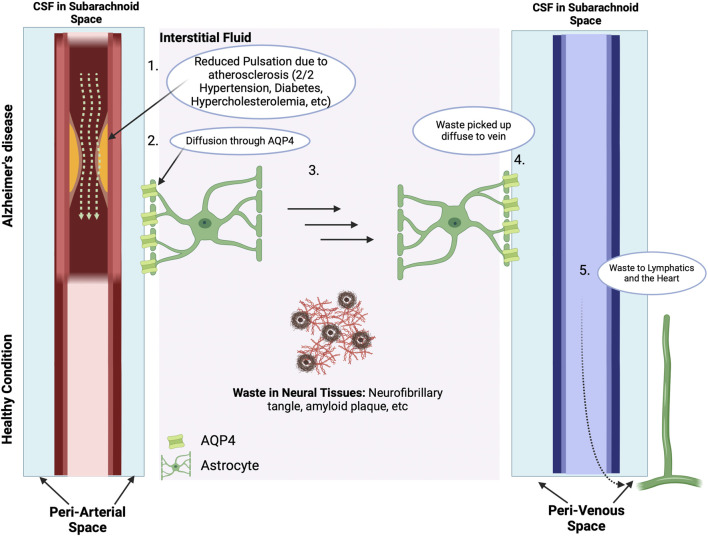
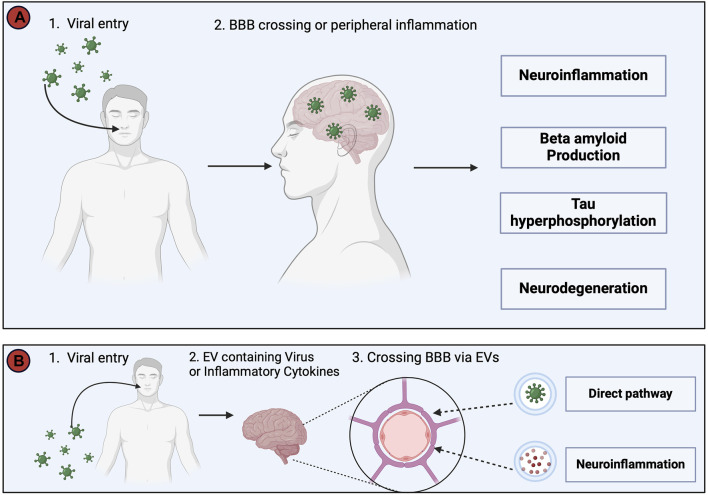
Alois Alzheimer described the first patient with Alzheimer's disease (AD) in 1907 and today AD is the most frequently diagnosed of dementias. AD is a multi-factorial neurodegenerative disorder with familial, life style and comorbidity influences impacting a global population of more than 47 million with a projected escalation by 2050 to exceed 130 million. In the USA the AD demographic encompasses approximately six million individuals, expected to increase to surpass 13 million by 2050, and the antecedent phase of AD, recognized as mild cognitive impairment (MCI), involves nearly 12 million individuals. The economic outlay for the management of AD and AD-related cognitive decline is estimated at approximately 355 billion USD. In addition, the intensifying prevalence of AD cases in countries with modest to intermediate income countries further enhances the urgency for more therapeutically and cost-effective treatments and for improving the quality of life for patients and their families. This narrative review evaluates the pathophysiological basis of AD with an initial focus on the therapeutic efficacy and limitations of the existing drugs that provide symptomatic relief: acetylcholinesterase inhibitors (AChEI) donepezil, galantamine, rivastigmine, and the N-methyl-D-aspartate receptor (NMDA) receptor allosteric modulator, memantine. The hypothesis that amyloid-β (Aβ) and tau are appropriate targets for drugs and have the potential to halt the progress of AD is critically analyzed with a particular focus on clinical trial data with anti-Aβ monoclonal antibodies (MABs), namely, aducanumab, lecanemab and donanemab. This review challenges the dogma that targeting Aβ will benefit the majority of subjects with AD that the anti-Aβ MABs are unlikely to be the "magic bullet". A comparison of the benefits and disadvantages of the different classes of drugs forms the basis for determining new directions for research and alternative drug targets that are undergoing pre-clinical and clinical assessments. In addition, we discuss and stress the importance of the treatment of the co-morbidities, including hypertension, diabetes, obesity and depression that are known to increase the risk of developing AD.
Keywords: Alzheimer’s disease; N-methyl-Daspartate receptor; acetylcholinesterase inhibitors; amyloid protein; donepezil; lecanemab; memantine; monoclonal antibody.
Copyright © 2024 Kim, Al Jerdi, MacDonald and Triggle.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Ackley S. F., Zimmerman S. C., Brenowitz W. D., Tchetgen E. J., Gold A. L., Manly J. J., et al. (2021). Effect of reductions in amyloid levels on cognitive change in randomized trials: instrumental variable meta-analysis. BMJ 372, n156. Erratum in: BMJ. 2022 Aug 30, 378, o2094. 10.1136/bmj.n156 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources