

FRET-guided modeling of nucleic acids
- PMID: 38869063
- PMCID: PMC11260485
- DOI: 10.1093/nar/gkae496
FRET-guided modeling of nucleic acids
Abstract
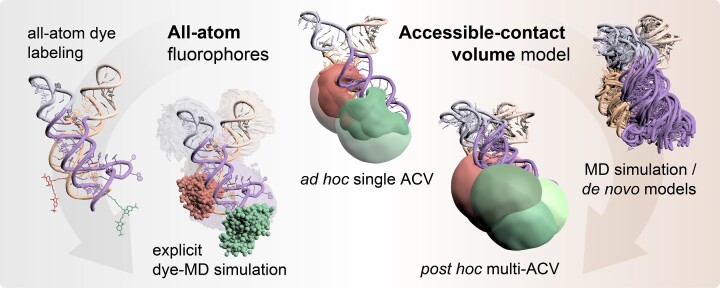
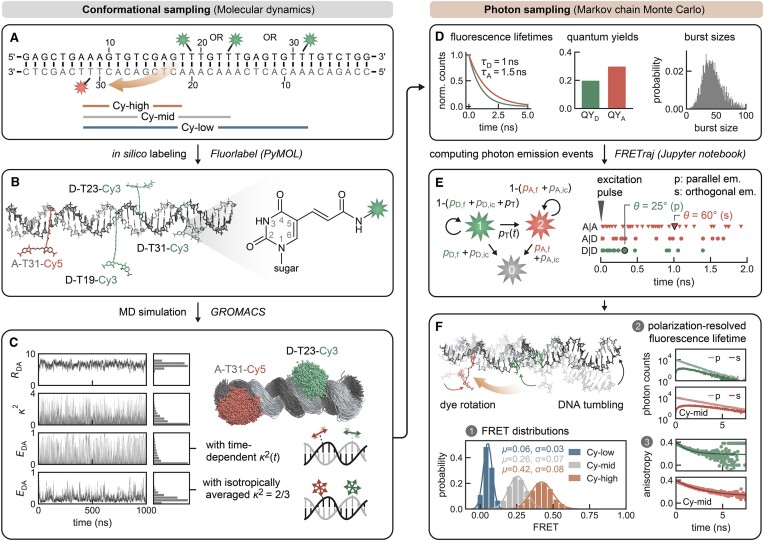
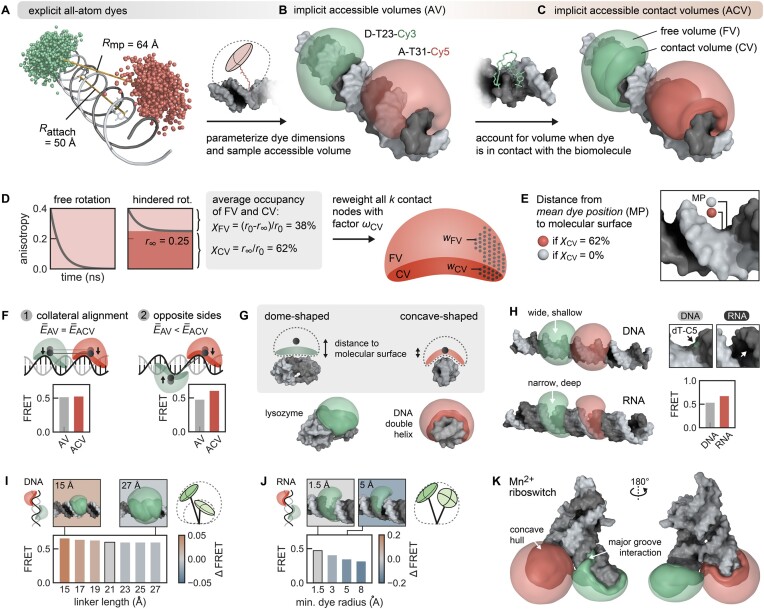
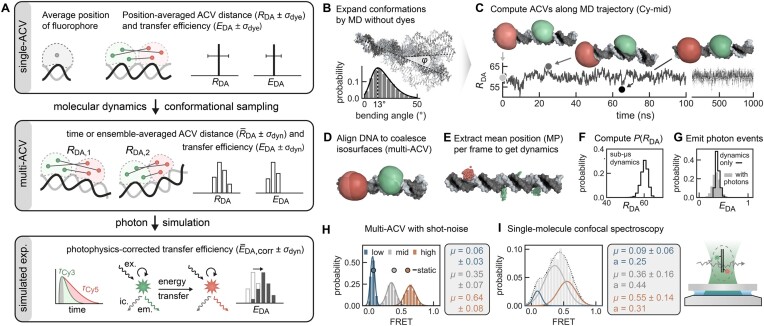
The functional diversity of RNAs is encoded in their innate conformational heterogeneity. The combination of single-molecule spectroscopy and computational modeling offers new attractive opportunities to map structural transitions within nucleic acid ensembles. Here, we describe a framework to harmonize single-molecule Förster resonance energy transfer (FRET) measurements with molecular dynamics simulations and de novo structure prediction. Using either all-atom or implicit fluorophore modeling, we recreate FRET experiments in silico, visualize the underlying structural dynamics and quantify the reaction coordinates. Using multiple accessible-contact volumes as a post hoc scoring method for fragment assembly in Rosetta, we demonstrate that FRET can be used to filter a de novo RNA structure prediction ensemble by refuting models that are not compatible with in vitro FRET measurement. We benchmark our FRET-assisted modeling approach on double-labeled DNA strands and validate it against an intrinsically dynamic manganese(II)-binding riboswitch. We show that a FRET coordinate describing the assembly of a four-way junction allows our pipeline to recapitulate the global fold of the riboswitch displayed by the crystal structure. We conclude that computational fluorescence spectroscopy facilitates the interpretability of dynamic structural ensembles and improves the mechanistic understanding of nucleic acid interactions.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Fluorogenic aptamers resolve the flexibility of RNA junctions using orientation-dependent FRET.RNA. 2021 Apr;27(4):433-444. doi: 10.1261/rna.078220.120. Epub 2020 Dec 29. RNA. 2021. PMID: 33376189 Free PMC article.
-
Observation of preQ1-II riboswitch dynamics using single-molecule FRET.RNA Biol. 2019 Sep;16(9):1086-1092. doi: 10.1080/15476286.2018.1536591. Epub 2018 Oct 30. RNA Biol. 2019. PMID: 30328747 Free PMC article.
-
Single-Molecule FRET Kinetics of the Mn2+ Riboswitch: Evidence for Allosteric Mg2+ Control of "Induced-Fit" vs "Conformational Selection" Folding Pathways.J Phys Chem B. 2019 Mar 7;123(9):2005-2015. doi: 10.1021/acs.jpcb.8b11841. Epub 2019 Feb 25. J Phys Chem B. 2019. PMID: 30739441
-
Multi-fluorophore fluorescence resonance energy transfer for probing nucleic acids structure and folding.Methods Mol Biol. 2006;335:257-71. doi: 10.1385/1-59745-069-3:257. Methods Mol Biol. 2006. PMID: 16785633 Review.
-
Using sm-FRET and denaturants to reveal folding landscapes.Methods Enzymol. 2014;549:313-41. doi: 10.1016/B978-0-12-801122-5.00014-3. Methods Enzymol. 2014. PMID: 25432755 Review.
Cited by
-
Analyzing aptamer structure and interactions: in silico modelling and instrumental methods.Biophys Rev. 2024 Nov 20;16(6):685-700. doi: 10.1007/s12551-024-01252-z. eCollection 2024 Dec. Biophys Rev. 2024. PMID: 39830127 Review.
-
A new twist on PIFE: photoisomerisation-related fluorescence enhancement.Methods Appl Fluoresc. 2023 Oct 12;12(1):012001. doi: 10.1088/2050-6120/acfb58. Methods Appl Fluoresc. 2023. PMID: 37726007 Free PMC article. Review.
References
-
- Bernetti M., Bussi G. Integrating experimental data with molecular simulations to investigate RNA structural dynamics. Curr. Opin. Struct. Biol. 2023; 78:102503. - PubMed
-
- Malhotra S., Träger S., Dal Peraro M., Topf M. Modelling structures in cryo-EM maps. Curr. Opin. Struct. Biol. 2019; 58:105–114. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
