

AMBRA1 levels predict resistance to MAPK inhibitors in melanoma
- PMID: 38870061
- PMCID: PMC11194594
- DOI: 10.1073/pnas.2400566121
AMBRA1 levels predict resistance to MAPK inhibitors in melanoma
Abstract
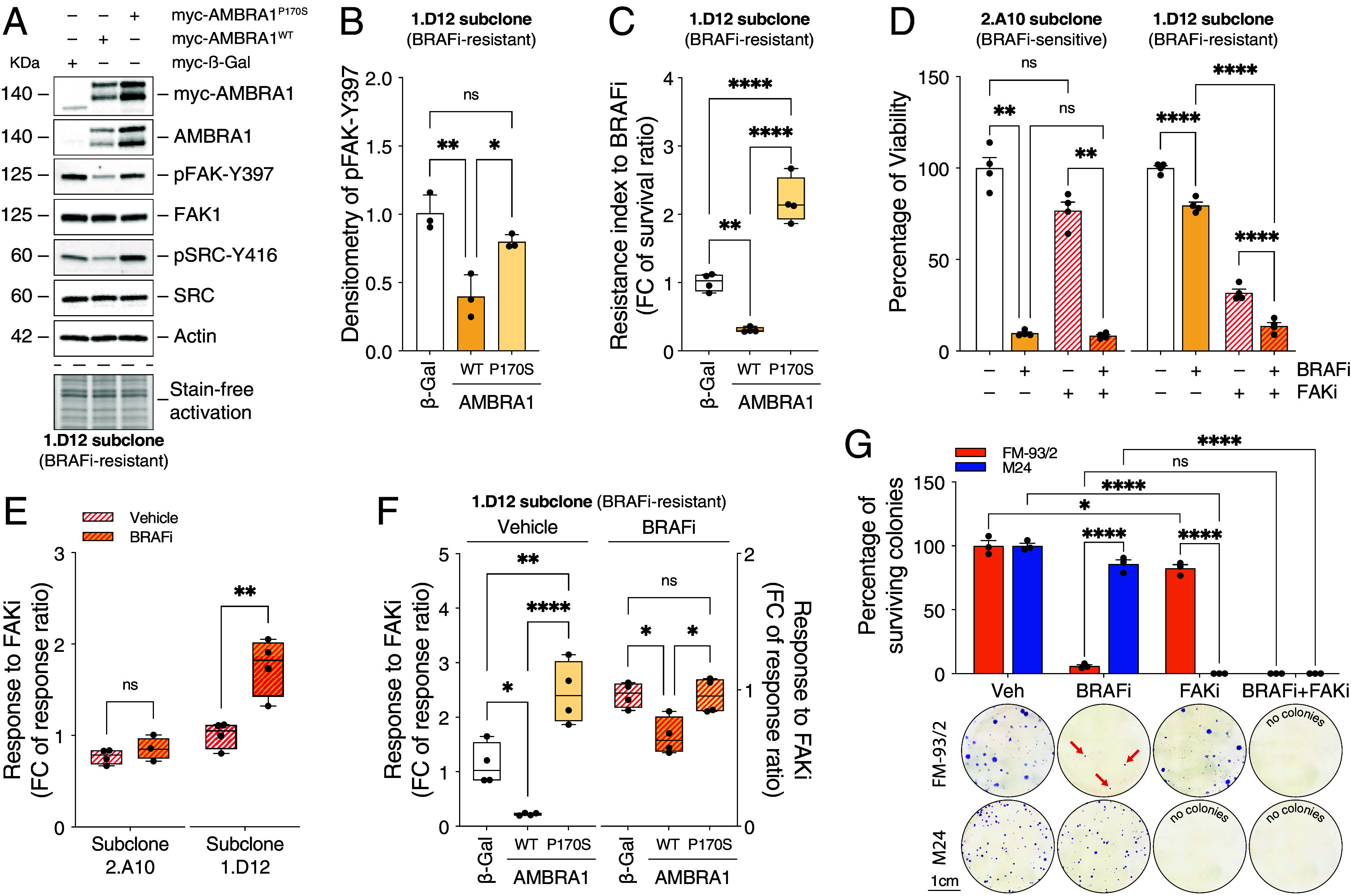
Intrinsic and acquired resistance to mitogen-activated protein kinase inhibitors (MAPKi) in melanoma remains a major therapeutic challenge. Here, we show that the clinical development of resistance to MAPKi is associated with reduced tumor expression of the melanoma suppressor Autophagy and Beclin 1 Regulator 1 (AMBRA1) and that lower expression levels of AMBRA1 predict a poor response to MAPKi treatment. Functional analyses show that loss of AMBRA1 induces phenotype switching and orchestrates an extracellular signal-regulated kinase (ERK)-independent resistance mechanism by activating focal adhesion kinase 1 (FAK1). In both in vitro and in vivo settings, melanomas with low AMBRA1 expression exhibit intrinsic resistance to MAPKi therapy but higher sensitivity to FAK1 inhibition. Finally, we show that the rapid development of resistance in initially MAPKi-sensitive melanomas can be attributed to preexisting subclones characterized by low AMBRA1 expression and that cotreatment with MAPKi and FAK1 inhibitors (FAKi) effectively prevents the development of resistance in these tumors. In summary, our findings underscore the value of AMBRA1 expression for predicting melanoma response to MAPKi and supporting the therapeutic efficacy of FAKi to overcome MAPKi-induced resistance.
Keywords: AMBRA1; FAK1; MAPK inhibitors; melanoma; targeted therapy.
Conflict of interest statement
Competing interests statement:P.L. is Chief Scientific Officer for AMLo Biosciences Ltd.
Figures







References
-
- Long G. V., Swetter S. M., Menzies A. M., Gershenwald J. E., Scolyer R. A., Cutaneous melanoma. Lancet 402, 485–502 (2023). - PubMed
-
- Shain A. H., Bastian B. C., From melanocytes to melanomas. Nat. Rev. Cancer 16, 345–358 (2016). - PubMed
-
- Ascierto P. A., et al. , Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 17, 1248–1260 (2016). - PubMed
MeSH terms
Substances
Grants and funding
- KBVU R204-A12424/Danish Cancer Society Research Center (DCRC)
- KBVU R352-A20515/Danish Cancer Society Research Center (DCRC)
- KBVU R231-A14034/Danish Cancer Society Research Center (DCRC)
- KBVU R325-A19075/Danish Cancer Society Research Center (DCRC)
- KBVU R146-A9414/Danish Cancer Society Research Center (DCRC)
- KBVU R231-A13855/Danish Cancer Society Research Center (DCRC)
- LF-OC-19-000004/LEO Fondet (LEO Foundation)
- LF-OC-19-000219/LEO Fondet (LEO Foundation)
- NNF18OC0052550/Novo Nordisk Fonden (NNF)
- NNF22OC0079352/Novo Nordisk Fonden (NNF)
- NNF21OC0070834/Novo Nordisk Fonden (NNF)
- IG2017-20719/Fondazione AIRC per la ricerca sul cancro ETS (AIRC)
- IG2019- 23543/Fondazione AIRC per la ricerca sul cancro ETS (AIRC)
- MRA 620385/Melanoma Research Alliance (MRA)
- NNF18SA0032928/Novo Nordisk Fonden (NNF)
- DNRF125/Danmarks Grundforskningsfond (DNRF)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
