

A phage tail-like bacteriocin suppresses competitors in metapopulations of pathogenic bacteria
- PMID: 38870284
- PMCID: PMC11404688
- DOI: 10.1126/science.ado0713
A phage tail-like bacteriocin suppresses competitors in metapopulations of pathogenic bacteria
Erratum in
-
Erratum for the Research Article "A phage tail-like bacteriocin suppresses competitors in metapopulations of pathogenic bacteria" by T. Backman et al.Science. 2025 Apr 11;388(6743):eadx9434. doi: 10.1126/science.adx9434. Epub 2025 Apr 10. Science. 2025. PMID: 40209003 No abstract available.
Abstract
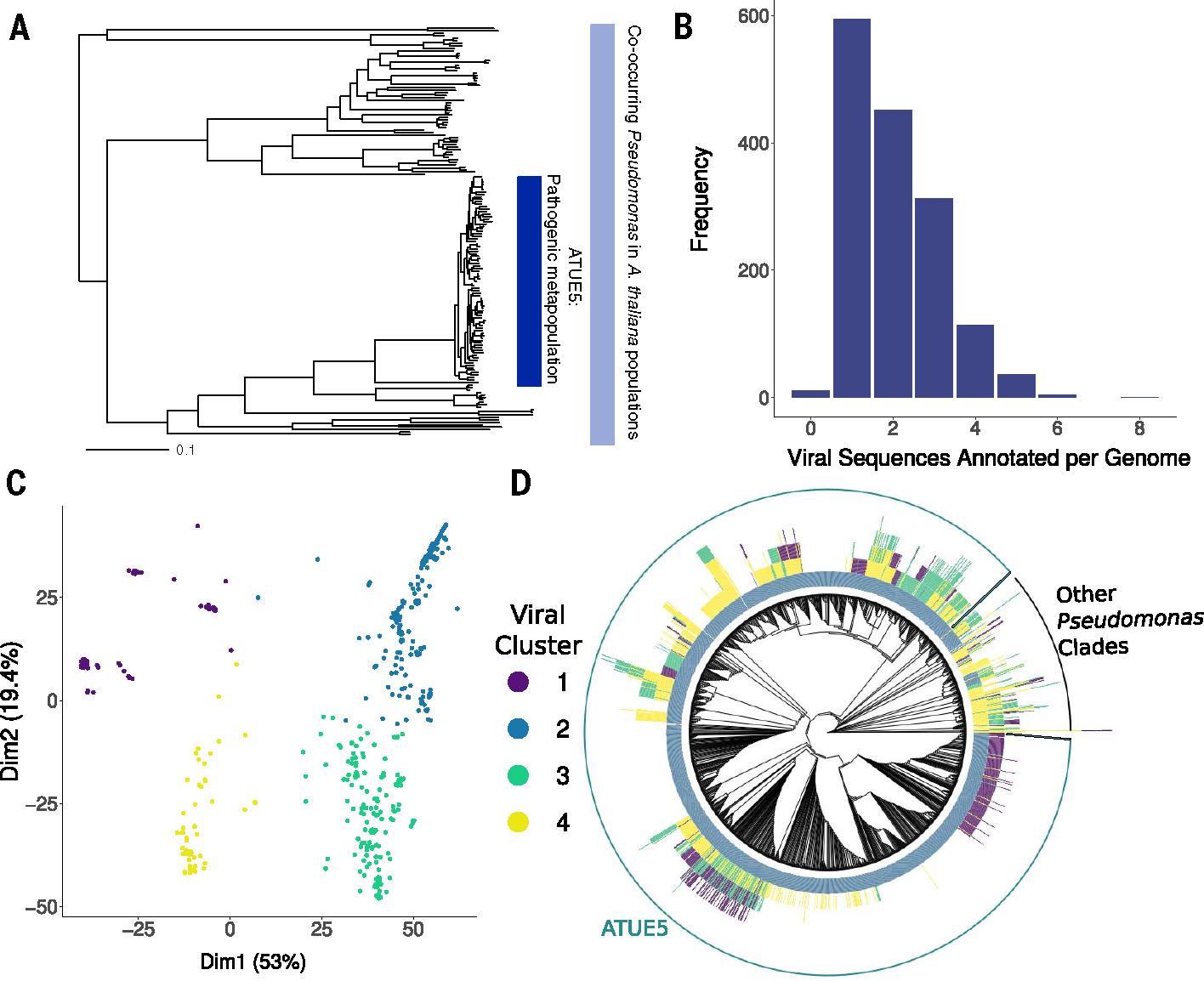
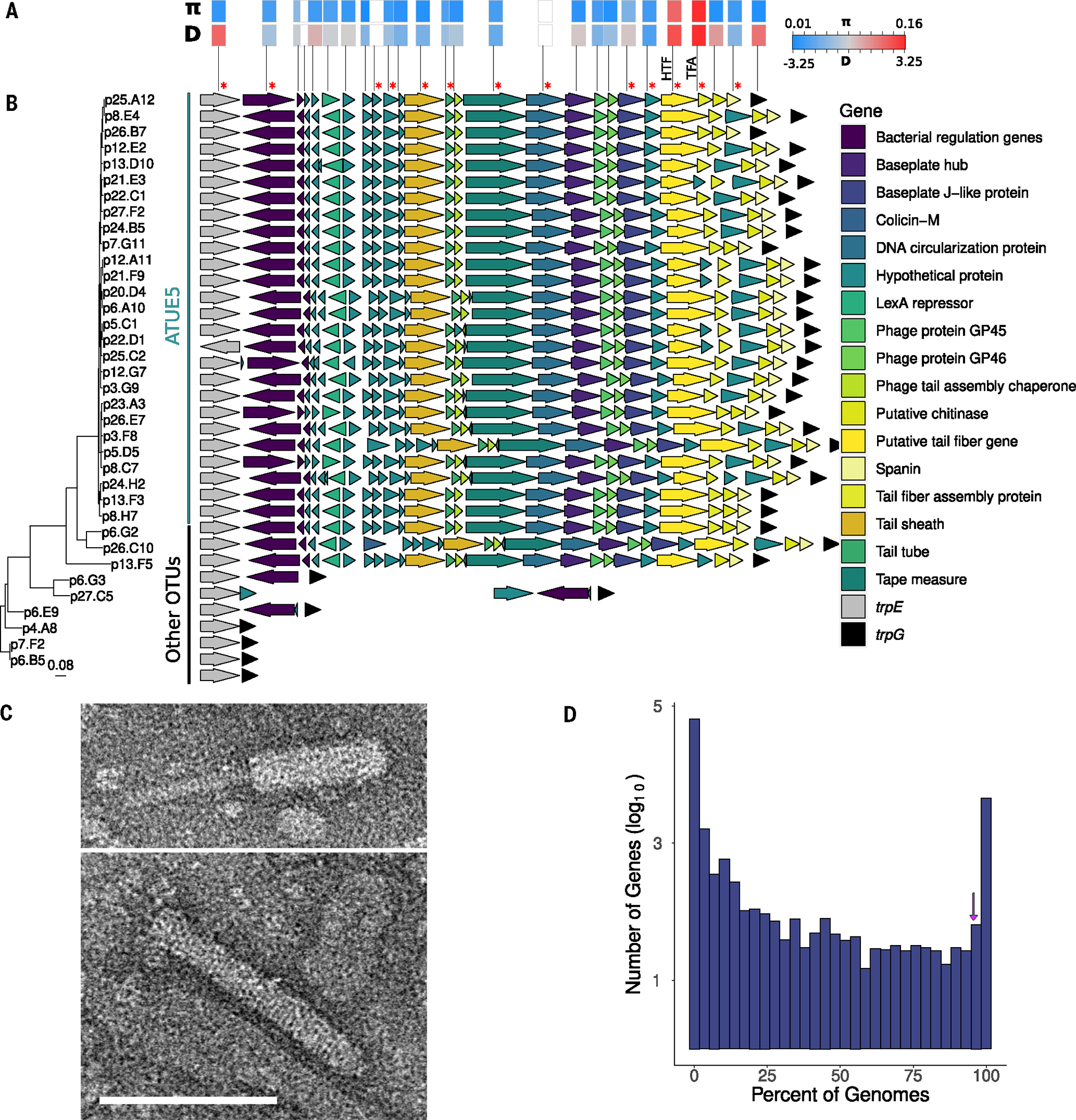
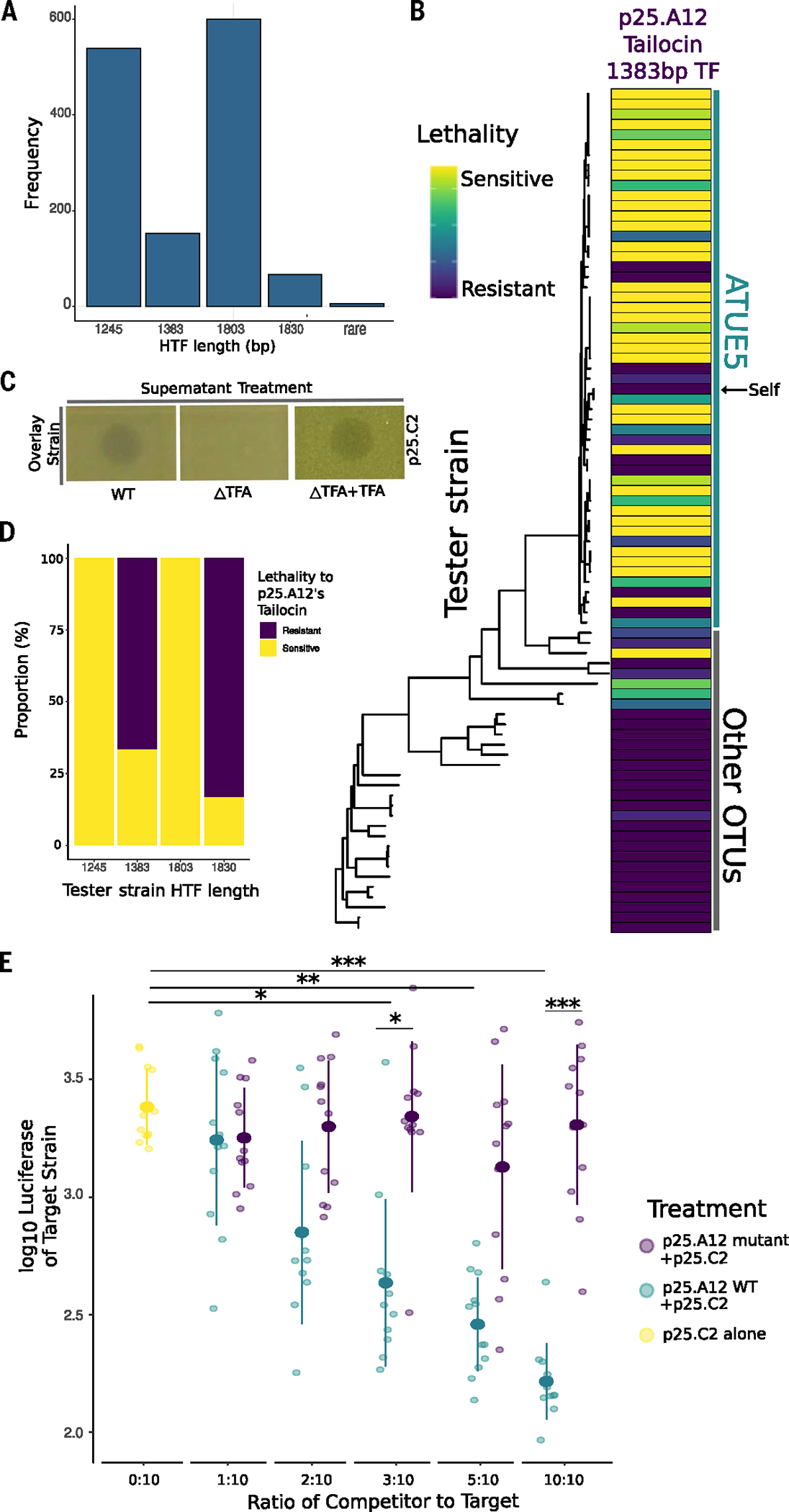
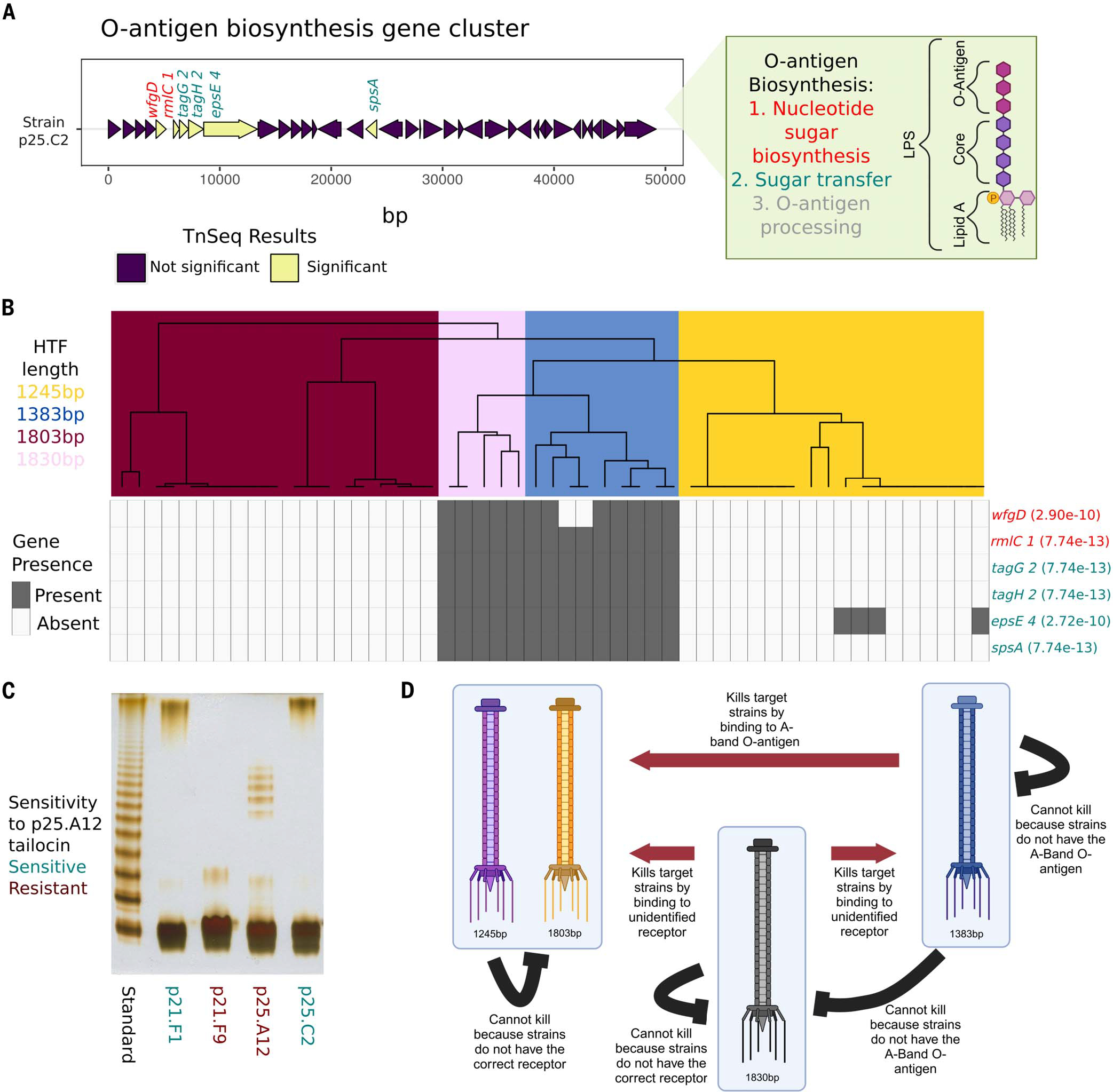
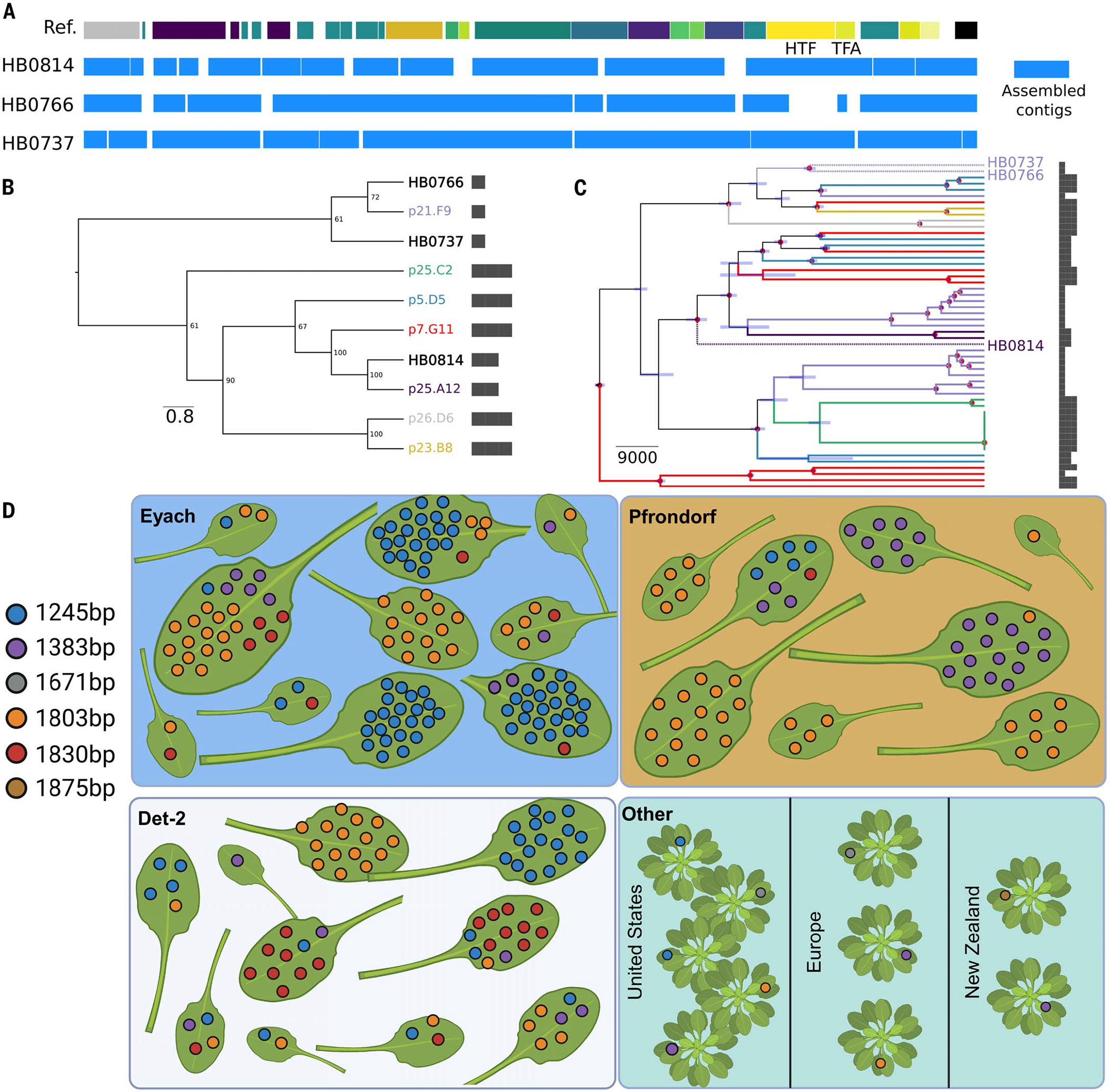
Bacteria can repurpose their own bacteriophage viruses (phage) to kill competing bacteria. Phage-derived elements are frequently strain specific in their killing activity, although there is limited evidence that this specificity drives bacterial population dynamics. Here, we identified intact phage and their derived elements in a metapopulation of wild plant-associated Pseudomonas genomes. We discovered that the most abundant viral cluster encodes a phage remnant resembling a phage tail called a tailocin, which bacteria have co-opted to kill bacterial competitors. Each pathogenic Pseudomonas strain carries one of a few distinct tailocin variants that target the variable polysaccharides in the outer membrane of co-occurring pathogenic Pseudomonas strains. Analysis of herbarium samples from the past 170 years revealed that the same tailocin and bacterial receptor variants have persisted in Pseudomonas populations. These results suggest that tailocin genetic diversity can be mined to develop targeted "tailocin cocktails" for microbial control.
Conflict of interest statement
Figures
Update of
-
A weaponized phage suppresses competitors in historical and modern metapopulations of pathogenic bacteria.bioRxiv [Preprint]. 2024 Feb 2:2023.04.17.536465. doi: 10.1101/2023.04.17.536465. bioRxiv. 2024. Update in: Science. 2024 Jun 14;384(6701):eado0713. doi: 10.1126/science.ado0713. PMID: 38352526 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
