

Integrative analysis of ultra-deep RNA-seq reveals alternative promoter usage as a mechanism of activating oncogenic programmes during prostate cancer progression
- PMID: 38871824
- PMCID: PMC11844022
- DOI: 10.1038/s41556-024-01438-3
Integrative analysis of ultra-deep RNA-seq reveals alternative promoter usage as a mechanism of activating oncogenic programmes during prostate cancer progression
Abstract
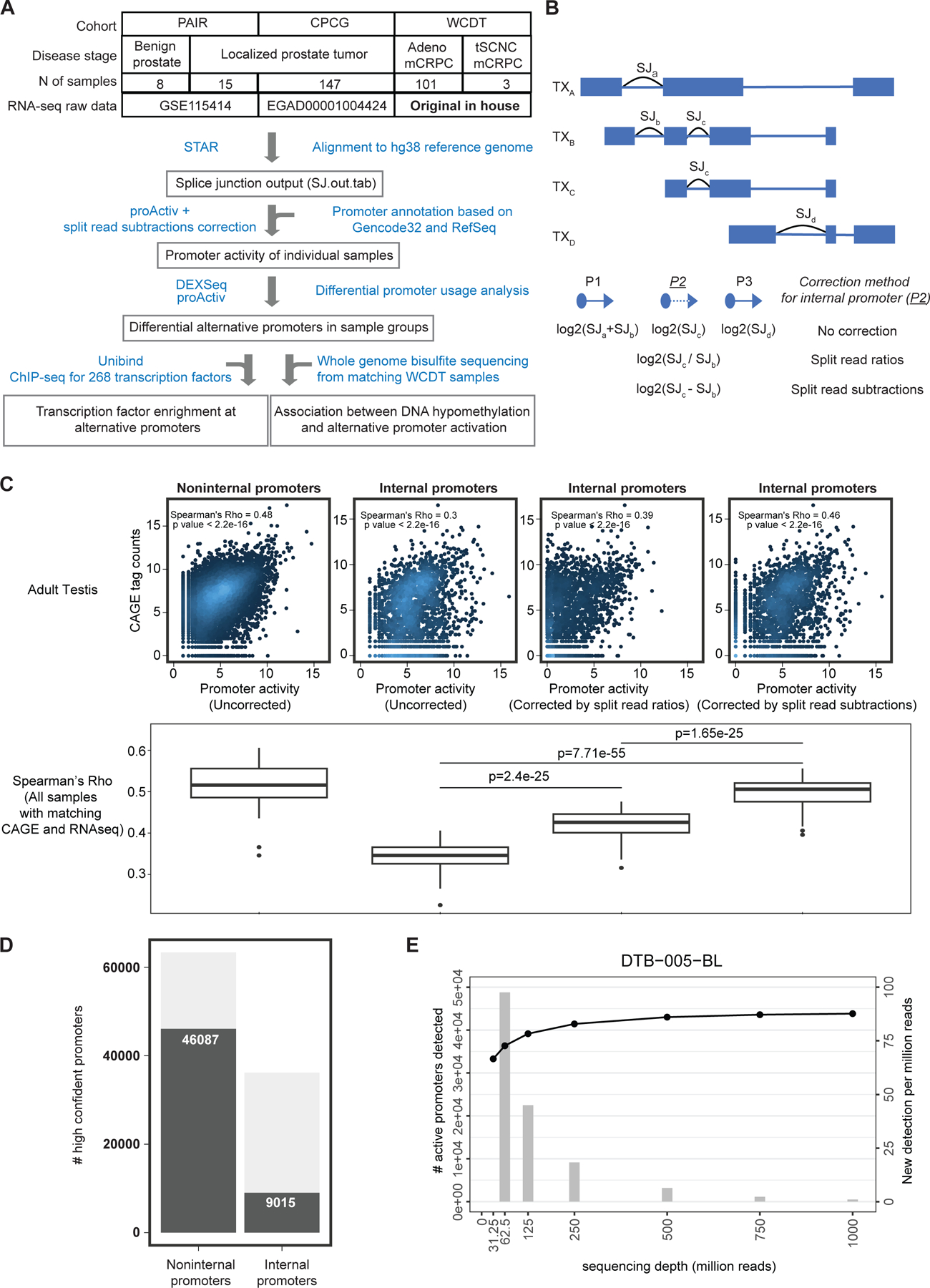
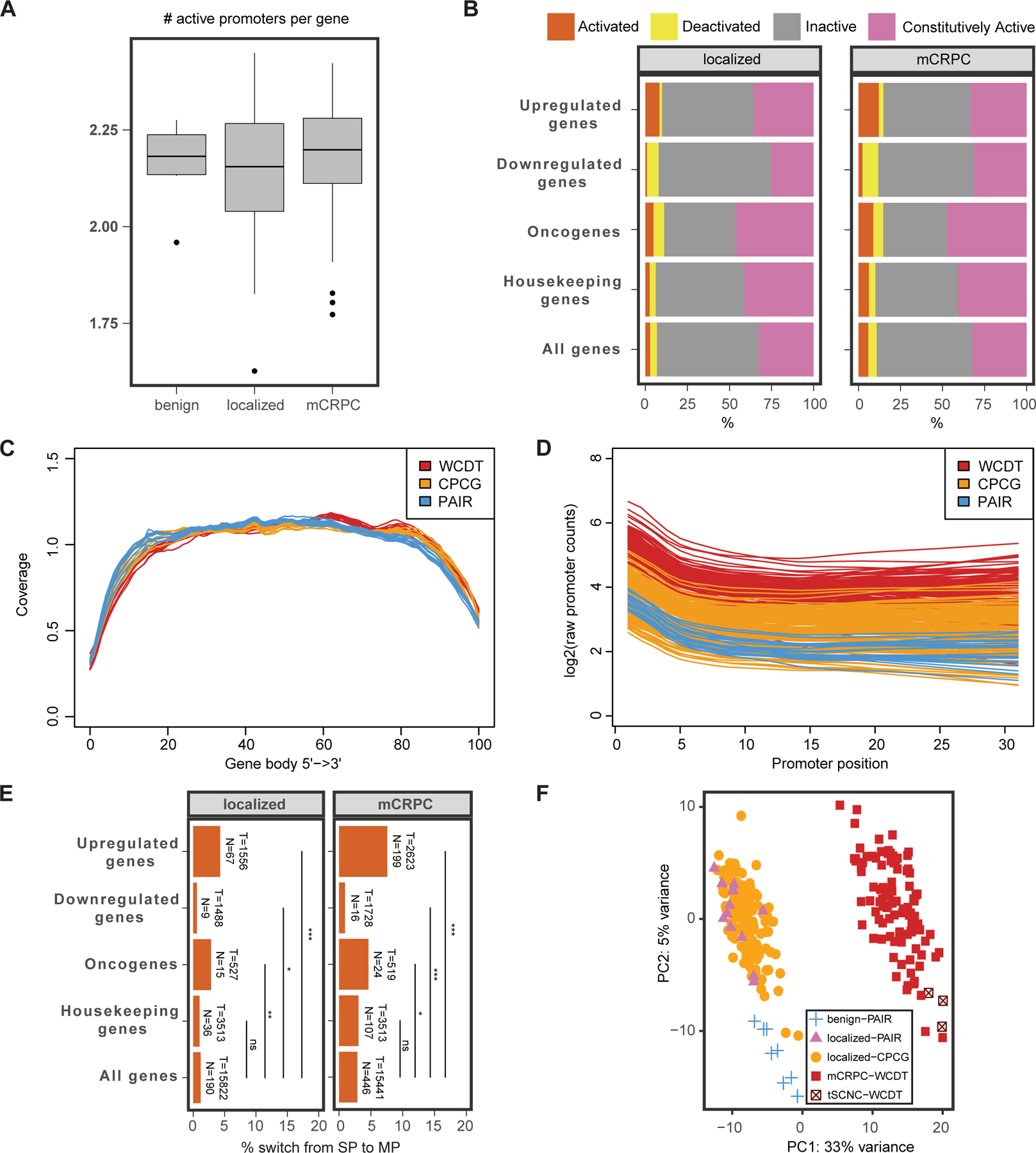
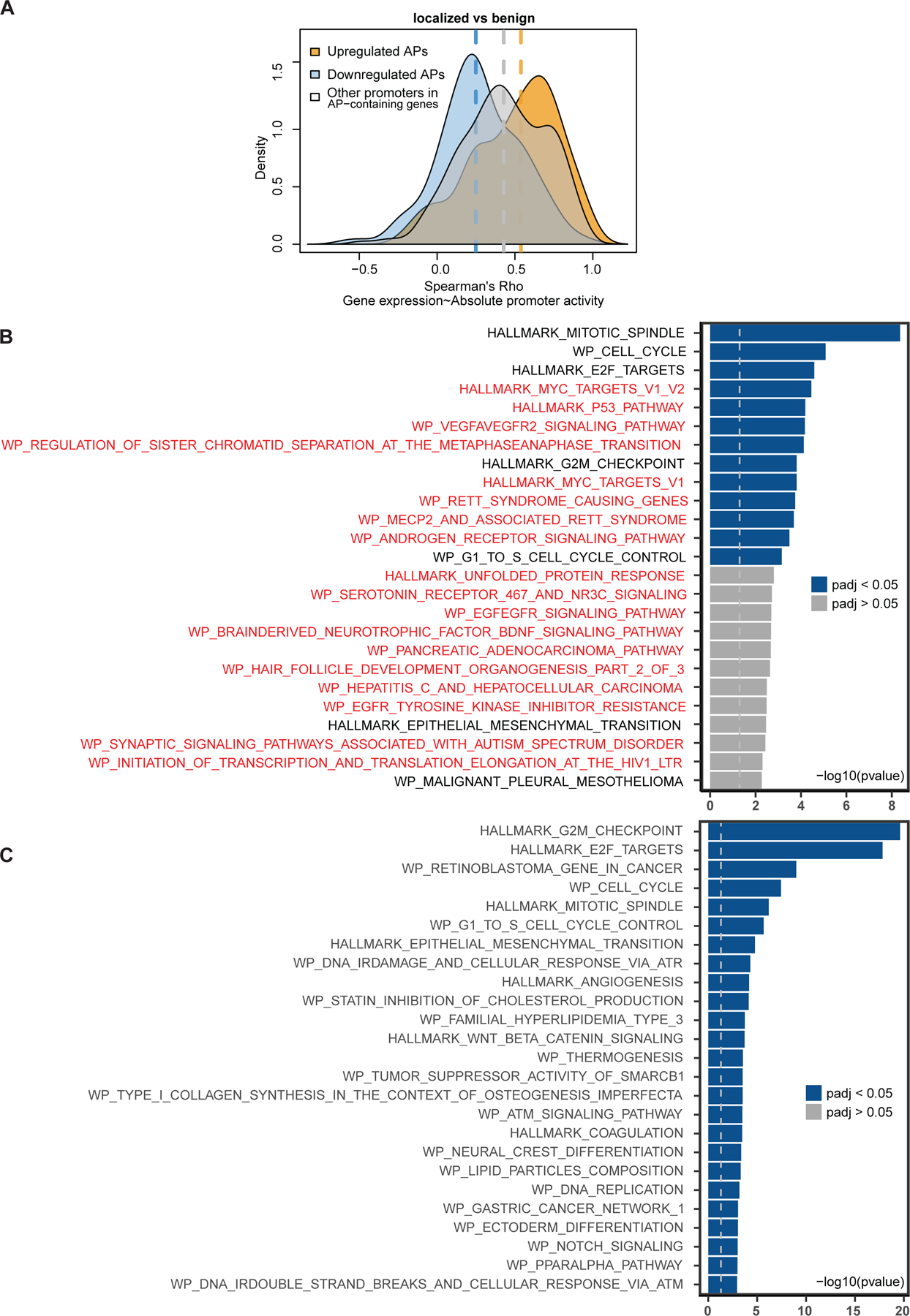
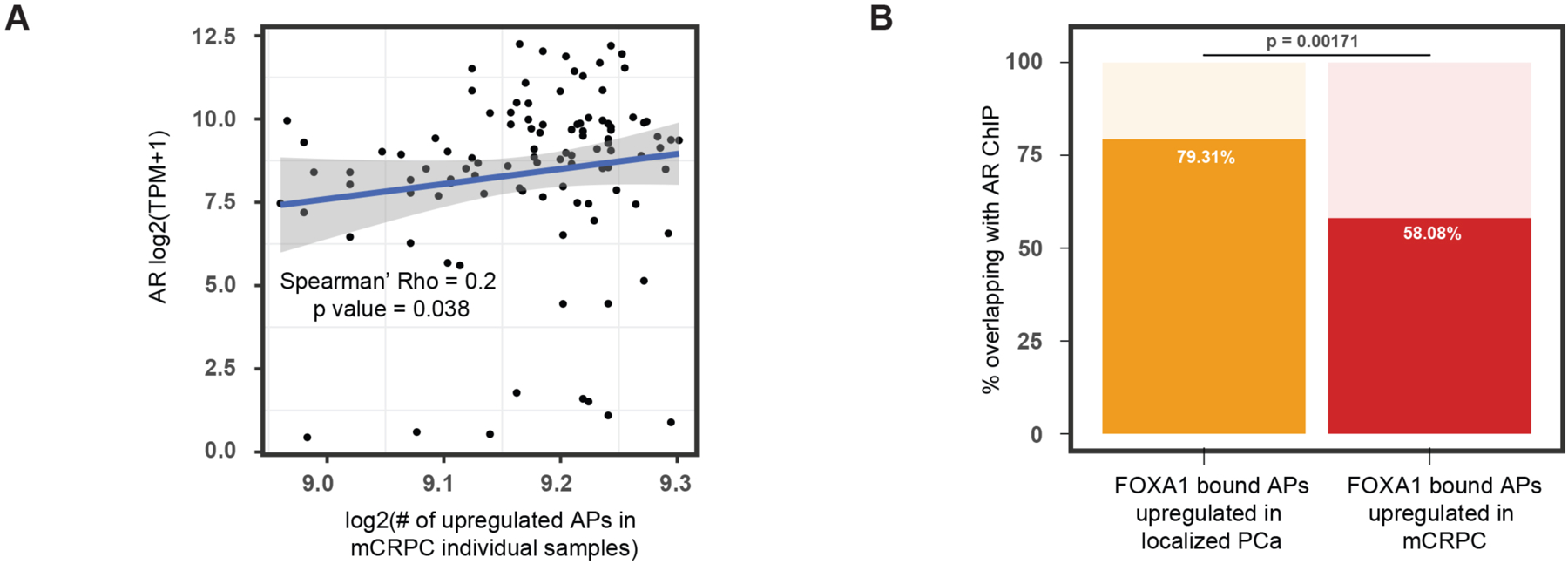
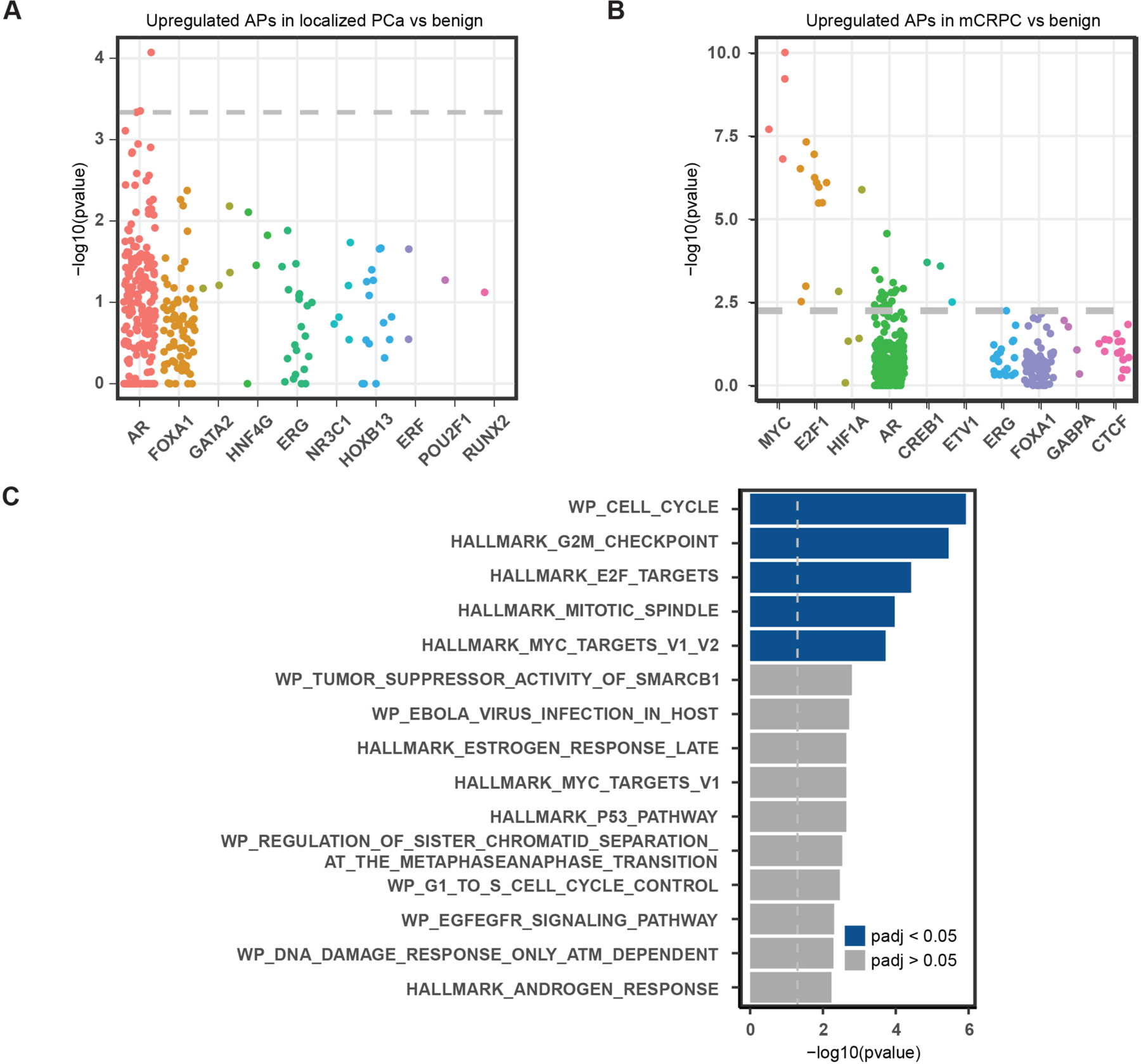
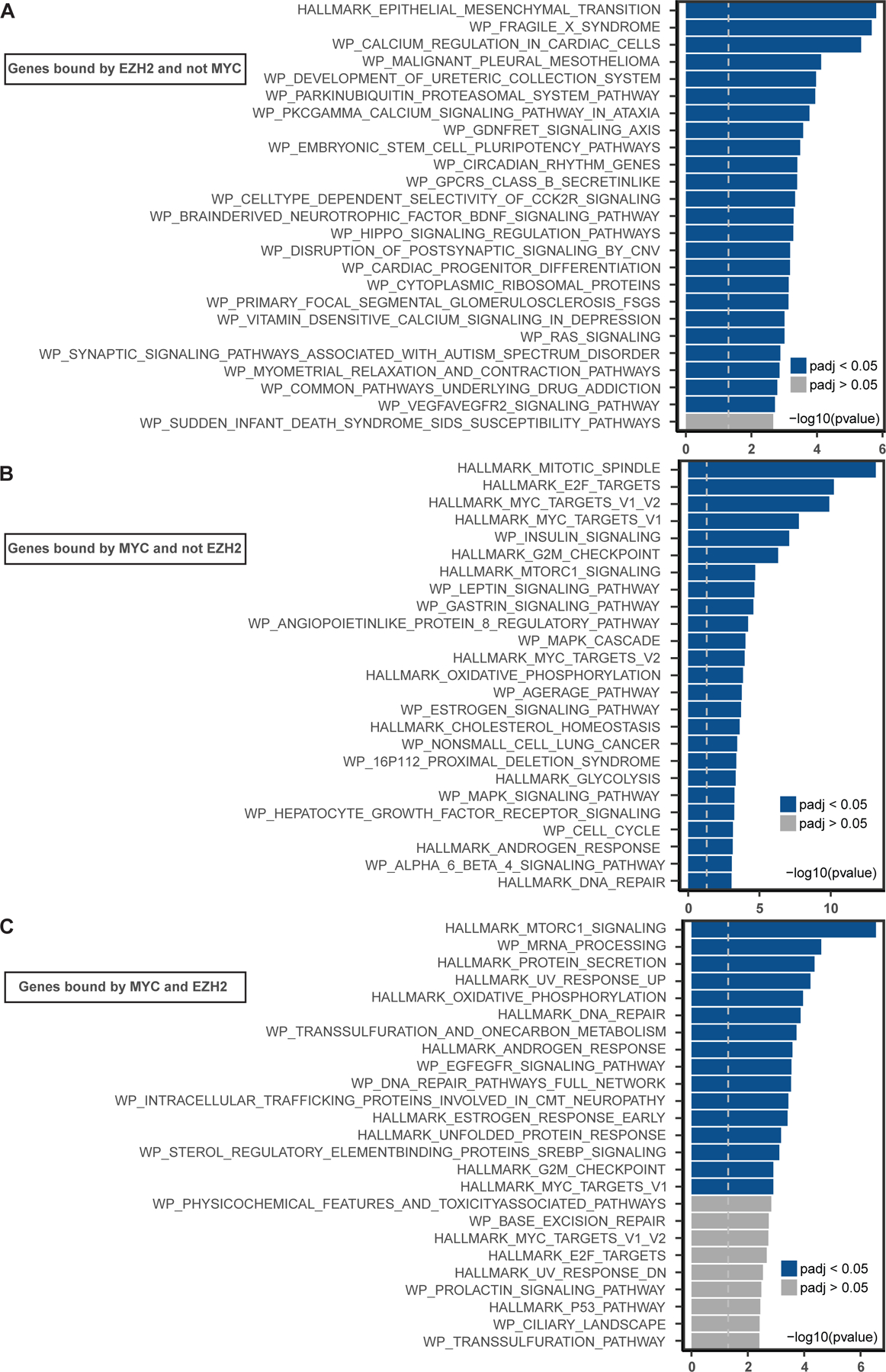
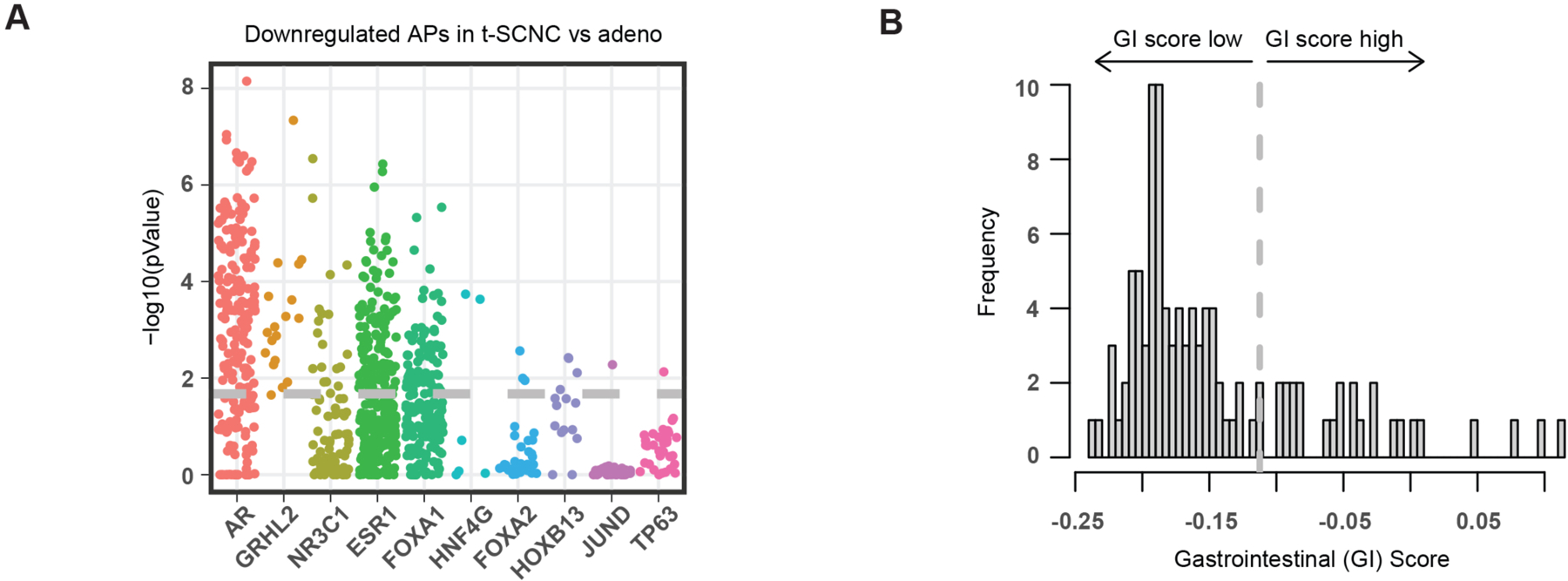
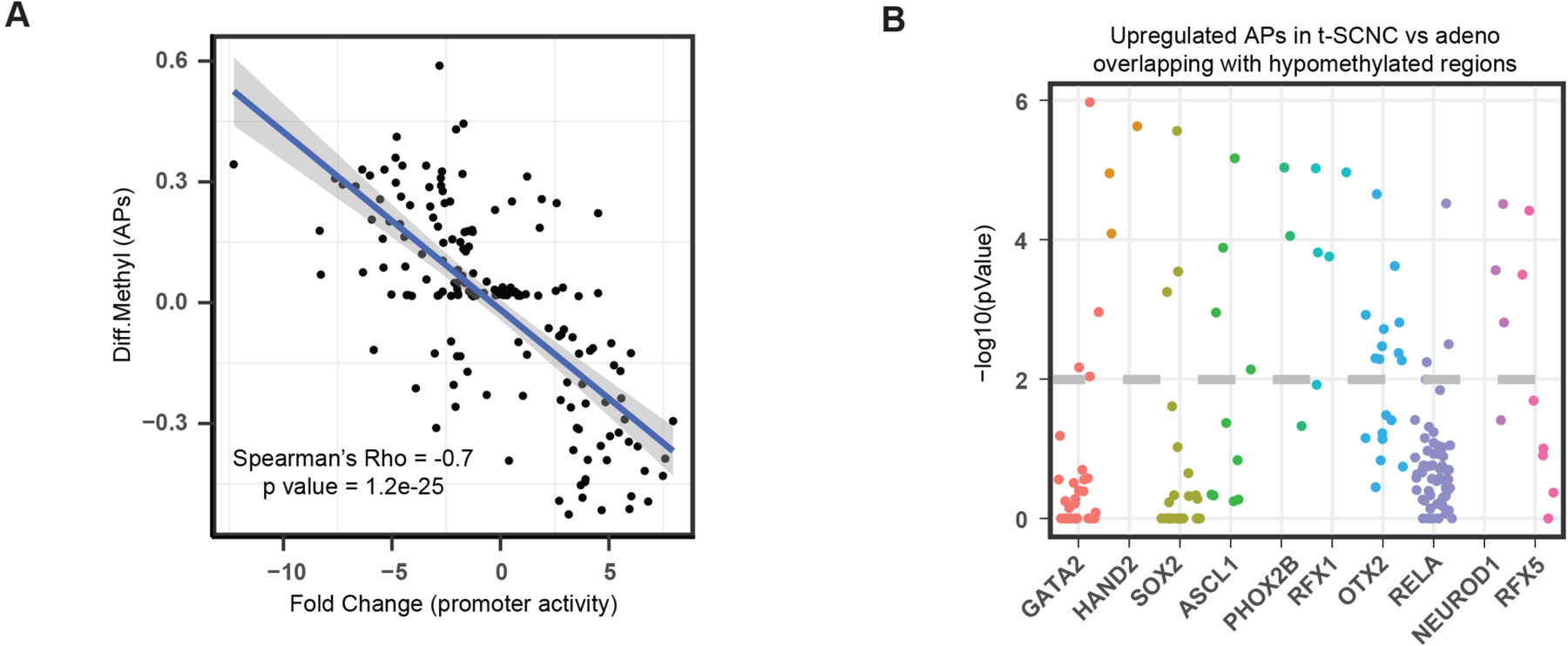
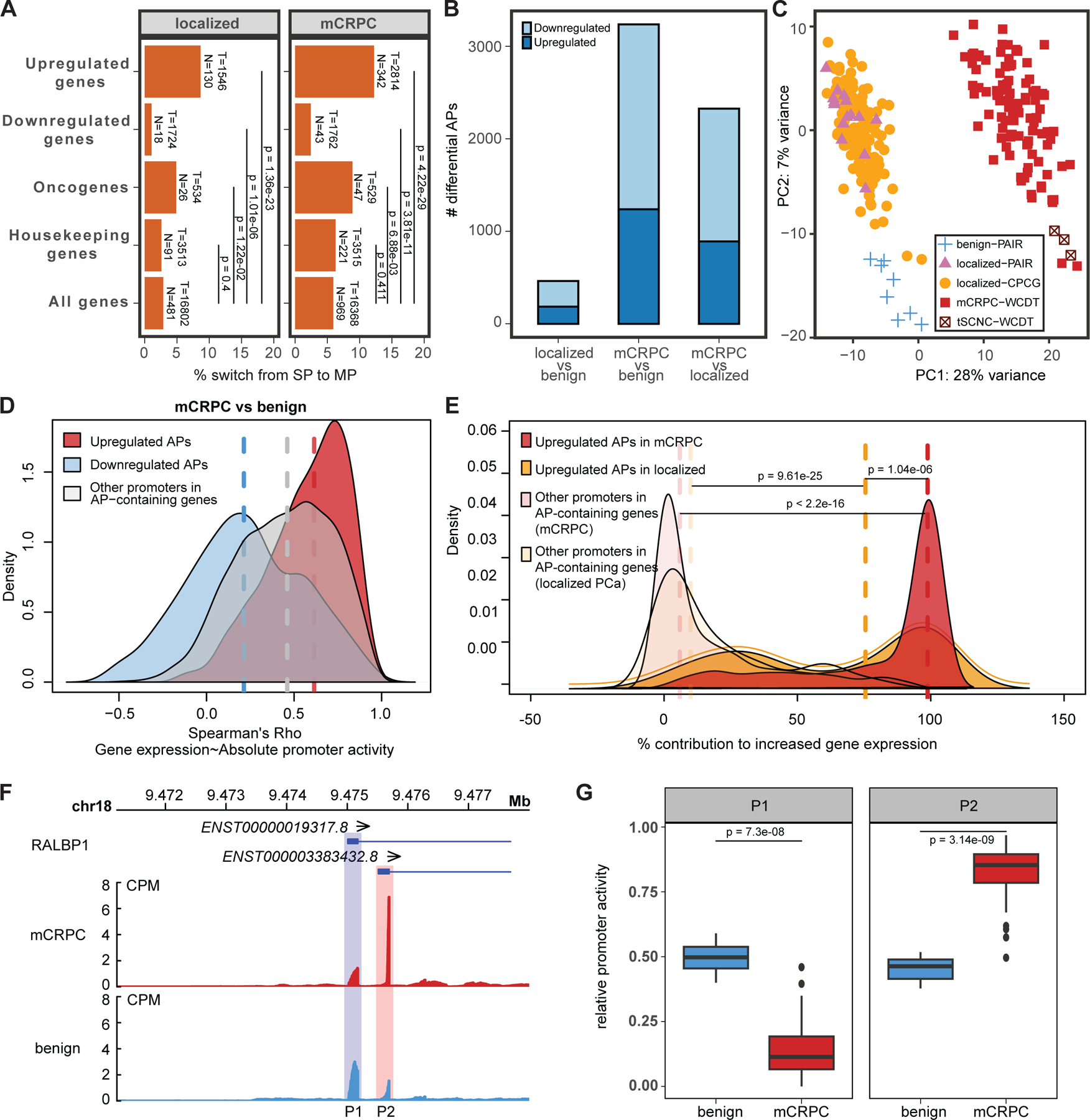
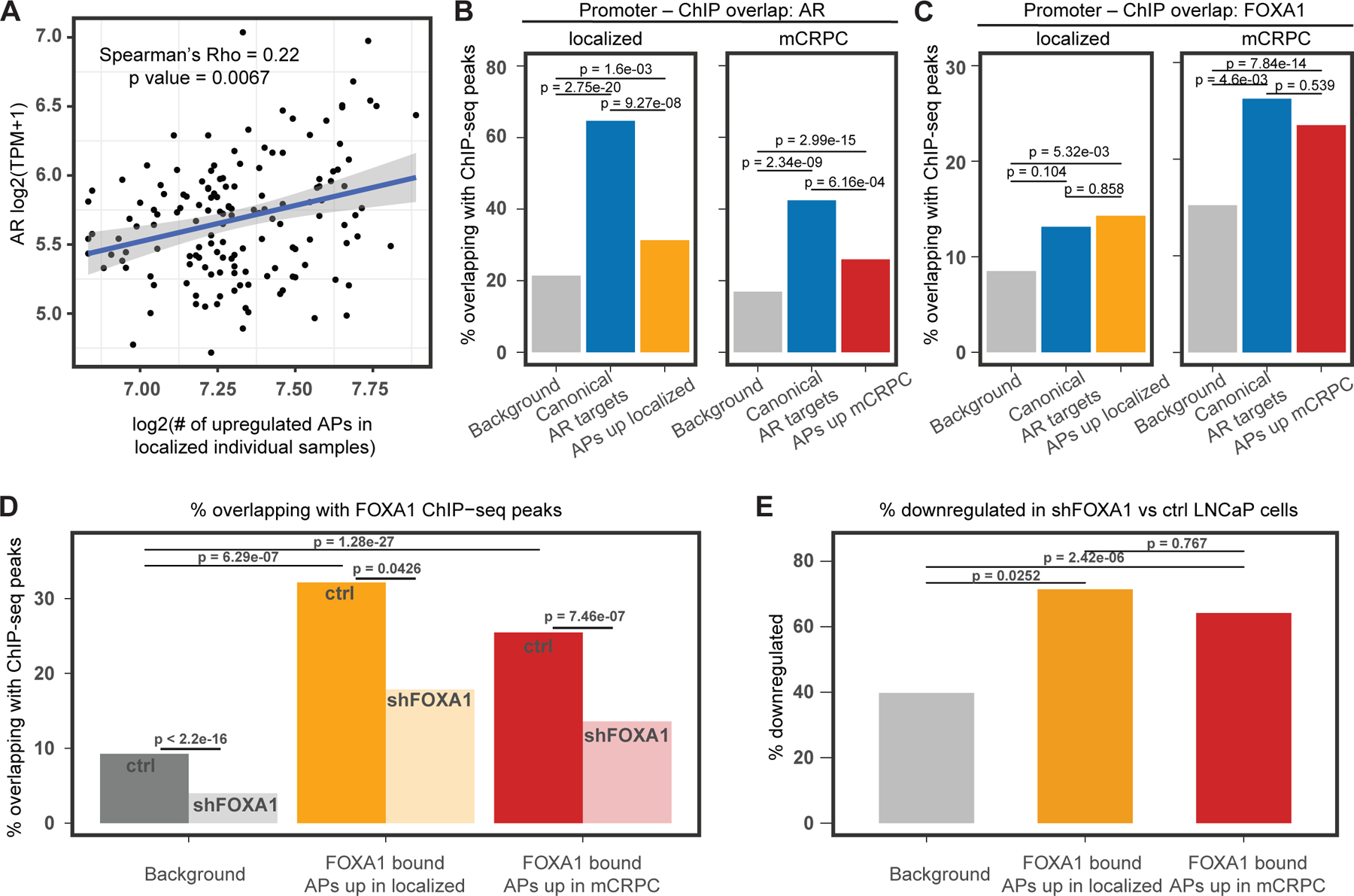
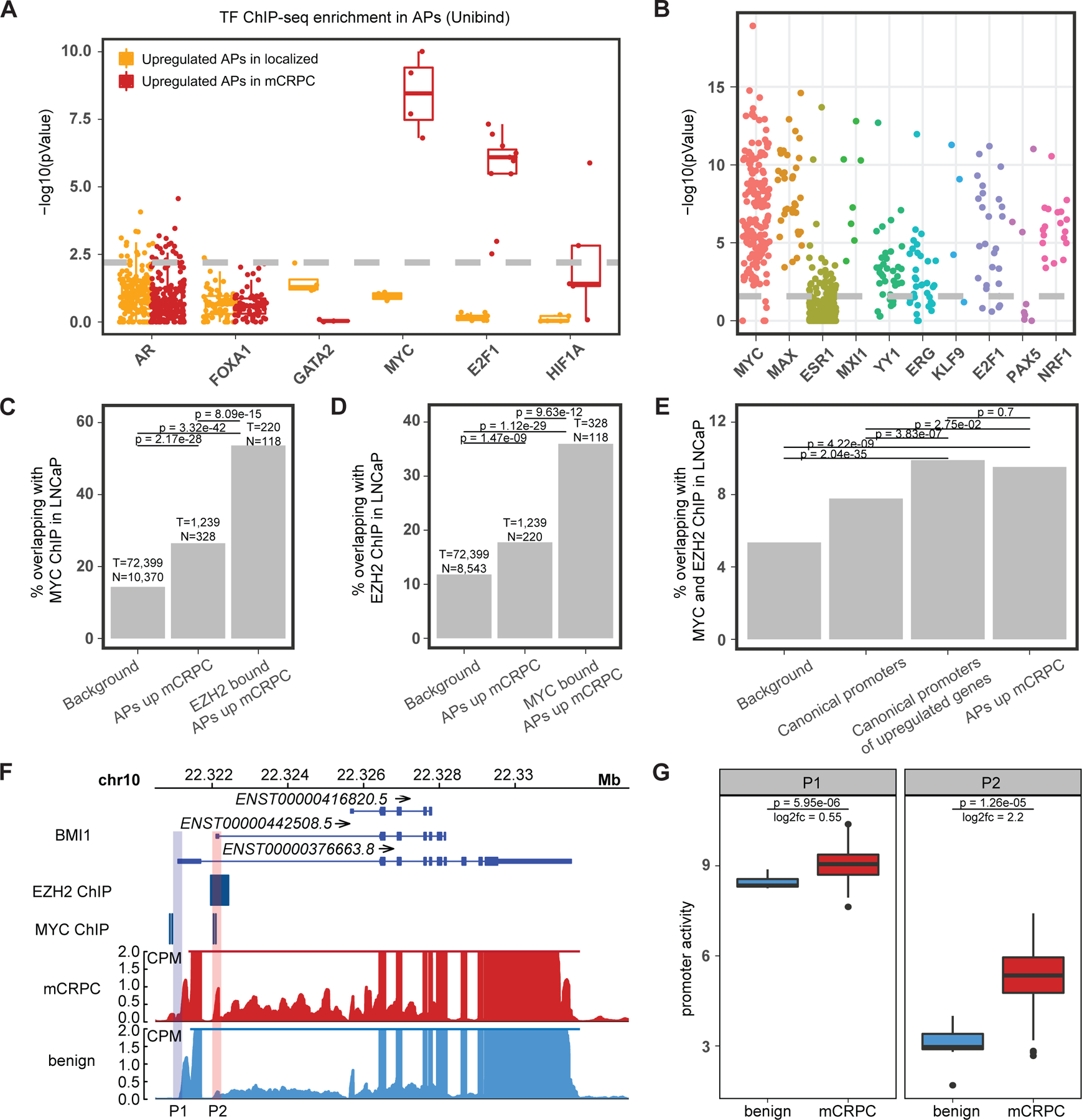
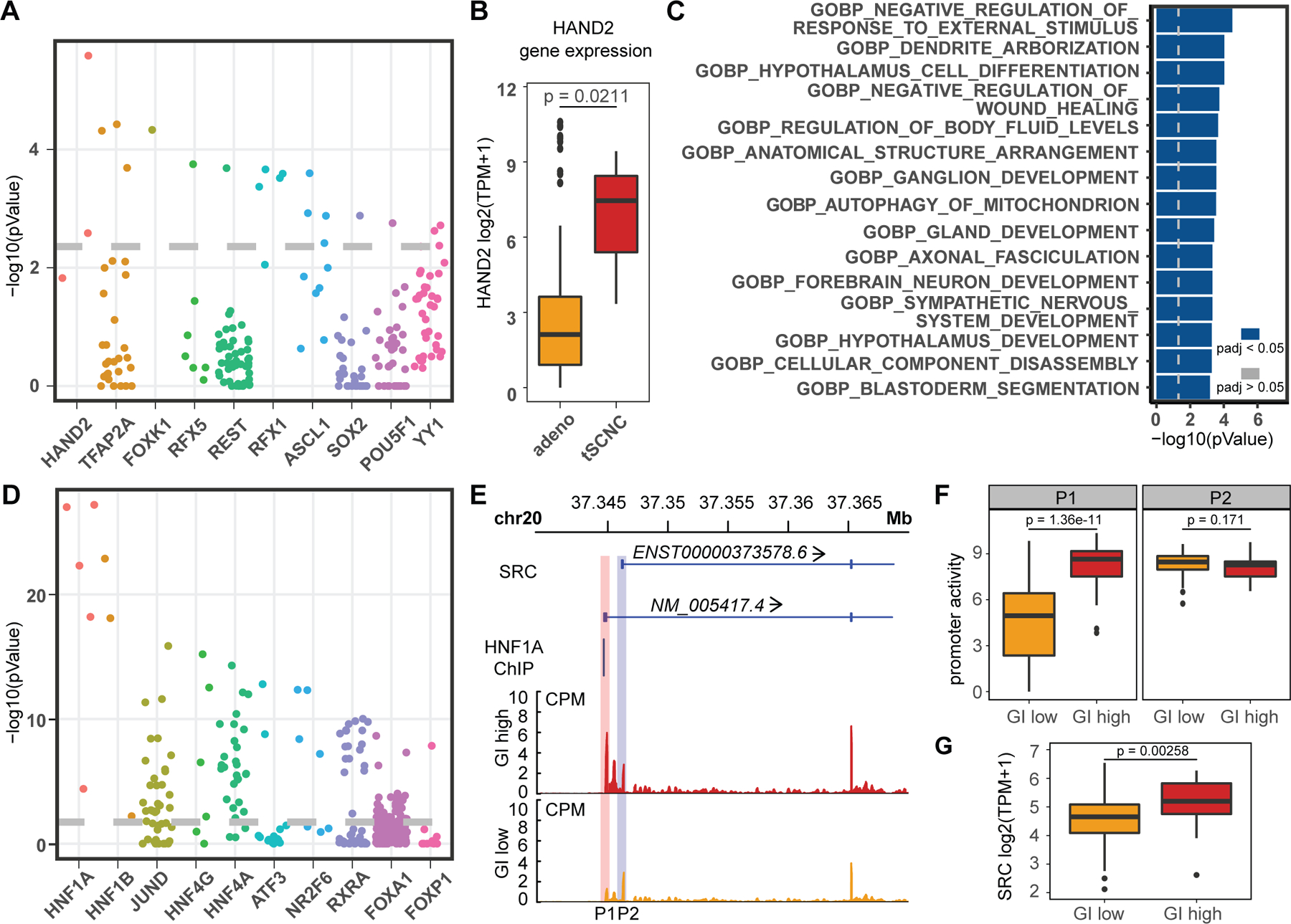
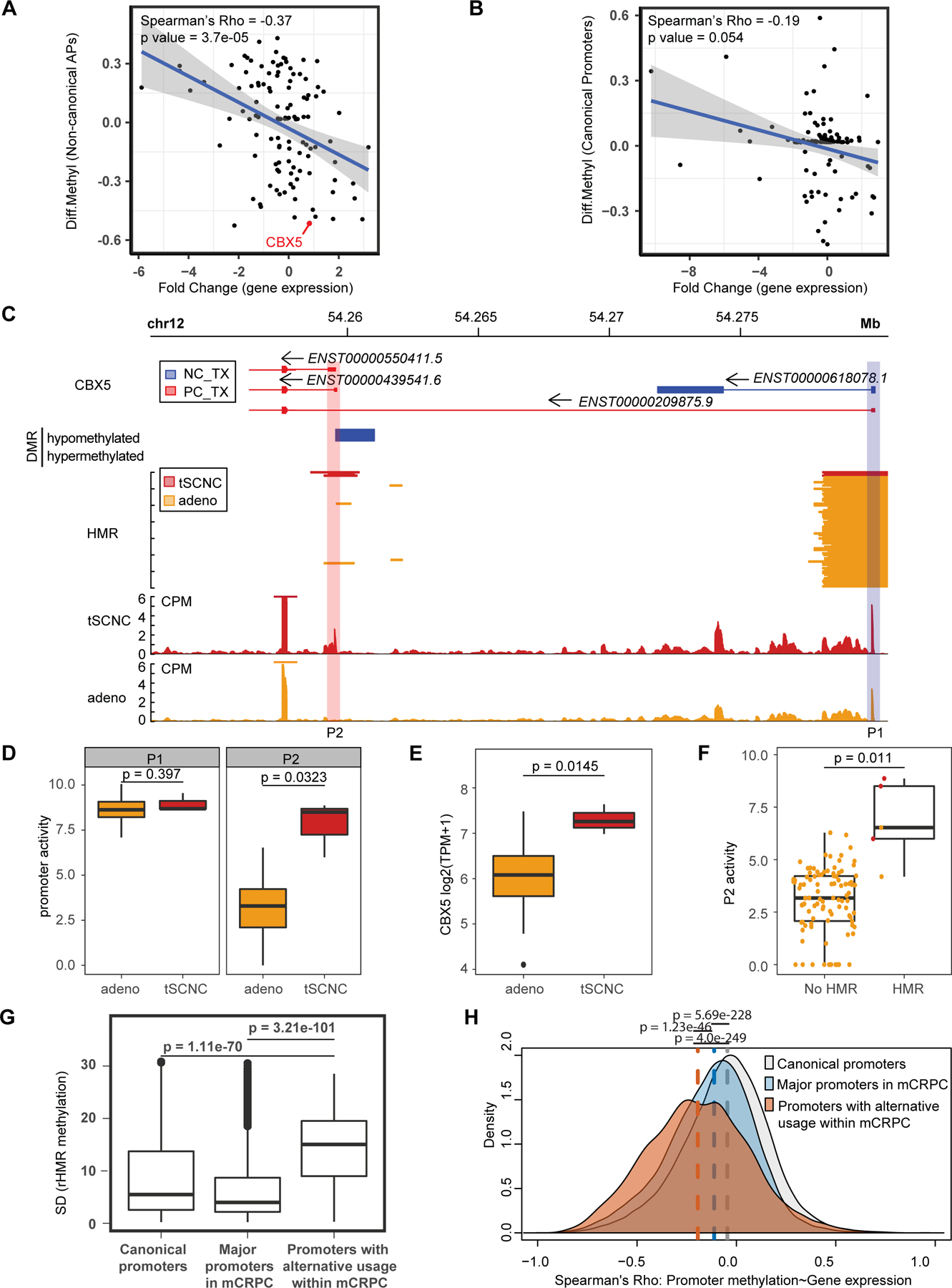
Transcription factor (TF) proteins regulate gene activity by binding to regulatory regions, most importantly at gene promoters. Many genes have alternative promoters (APs) bound by distinct TFs. The role of differential TF activity at APs during tumour development is poorly understood. Here we show, using deep RNA sequencing in 274 biopsies of benign prostate tissue, localized prostate tumours and metastatic castration-resistant prostate cancer, that AP usage increases as tumours progress and APs are responsible for a disproportionate amount of tumour transcriptional activity. Expression of the androgen receptor (AR), the key driver of prostate tumour activity, is correlated with elevated AP usage. We identified AR, FOXA1 and MYC as potential drivers of AP activation. DNA methylation is a likely mechanism for AP activation during tumour progression and lineage plasticity. Our data suggest that prostate tumours activate APs to magnify the transcriptional impact of tumour drivers, including AR and MYC.
© 2024. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
COMPETING INTERESTS
J.J.A. has consulted for or held advisory roles at Astellas Pharma, Bayer and Janssen Biotech Inc. He has received research funding from Aragon Pharmaceuticals Inc., Astellas Pharma, Novartis, Zenith Epigenetics Ltd. and Gilead Sciences Inc. F.Y.F. has consulted for Astellas, Bayer, Blue Earth Diagnostics, BMS, EMD Serono, Exact Sciences, Foundation Medicine, Janssen Oncology, Myovant, Roivant, and Varian, and serves on the Scientific Advisory Board for BlueStar Genomics and SerImmune. F.Y.F. has patent applications with Decipher Biosciences on molecular signatures in prostate cancer unrelated to this work. F.Y.F. has a patent application licensed to PFS Genomics/Exact Sciences. F.Y.F. has patent applications with Celgene unrelated to this work. The remaining authors declare no competing interests.
Figures
Comment in
-
Alternative promoters activate oncogenic programmes.Nat Rev Urol. 2024 Aug;21(8):456. doi: 10.1038/s41585-024-00919-2. Nat Rev Urol. 2024. PMID: 38997501 No abstract available.
Similar articles
-
USP11 promotes prostate cancer progression by up-regulating AR and c-Myc activity.Proc Natl Acad Sci U S A. 2024 Jul 30;121(31):e2403331121. doi: 10.1073/pnas.2403331121. Epub 2024 Jul 25. Proc Natl Acad Sci U S A. 2024. PMID: 39052835 Free PMC article.
-
Selective targeting of PARP-2 inhibits androgen receptor signaling and prostate cancer growth through disruption of FOXA1 function.Proc Natl Acad Sci U S A. 2019 Jul 16;116(29):14573-14582. doi: 10.1073/pnas.1908547116. Epub 2019 Jul 2. Proc Natl Acad Sci U S A. 2019. PMID: 31266892 Free PMC article.
-
Elevated levels of FOXA1 facilitate androgen receptor chromatin binding resulting in a CRPC-like phenotype.Oncogene. 2014 Dec 11;33(50):5666-74. doi: 10.1038/onc.2013.508. Epub 2013 Dec 2. Oncogene. 2014. PMID: 24292680 Free PMC article.
-
Transcriptional Regulation in Prostate Cancer.Cold Spring Harb Perspect Med. 2018 Nov 1;8(11):a030437. doi: 10.1101/cshperspect.a030437. Cold Spring Harb Perspect Med. 2018. PMID: 29530947 Free PMC article. Review.
-
Shaping Chromatin States in Prostate Cancer by Pioneer Transcription Factors.Cancer Res. 2020 Jun 15;80(12):2427-2436. doi: 10.1158/0008-5472.CAN-19-3447. Epub 2020 Feb 24. Cancer Res. 2020. PMID: 32094298 Free PMC article. Review.
Cited by
-
Multi-omics analysis identifies DLX4 as a novel biomarker for diagnosis, prognosis, and immune infiltration: from pan-cancer to renal cancer.Discov Oncol. 2025 Apr 5;16(1):467. doi: 10.1007/s12672-025-02258-z. Discov Oncol. 2025. PMID: 40186710 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
