

CellRank 2: unified fate mapping in multiview single-cell data
- PMID: 38871986
- PMCID: PMC11239496
- DOI: 10.1038/s41592-024-02303-9
CellRank 2: unified fate mapping in multiview single-cell data
Abstract
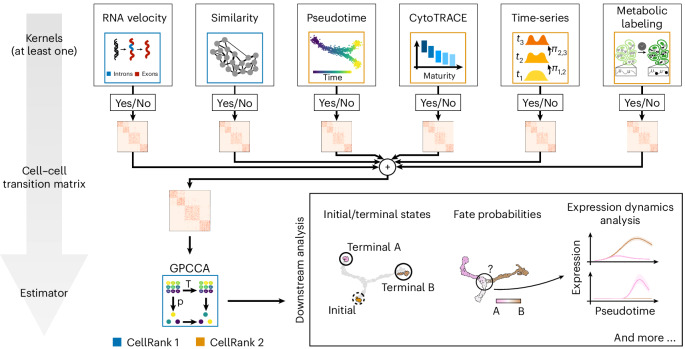
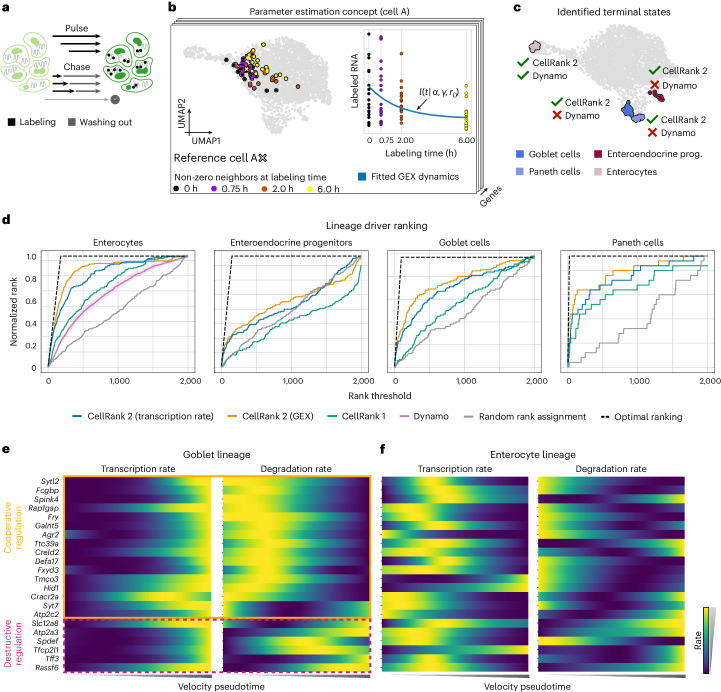
Single-cell RNA sequencing allows us to model cellular state dynamics and fate decisions using expression similarity or RNA velocity to reconstruct state-change trajectories; however, trajectory inference does not incorporate valuable time point information or utilize additional modalities, whereas methods that address these different data views cannot be combined or do not scale. Here we present CellRank 2, a versatile and scalable framework to study cellular fate using multiview single-cell data of up to millions of cells in a unified fashion. CellRank 2 consistently recovers terminal states and fate probabilities across data modalities in human hematopoiesis and endodermal development. Our framework also allows combining transitions within and across experimental time points, a feature we use to recover genes promoting medullary thymic epithelial cell formation during pharyngeal endoderm development. Moreover, we enable estimating cell-specific transcription and degradation rates from metabolic-labeling data, which we apply to an intestinal organoid system to delineate differentiation trajectories and pinpoint regulatory strategies.
© 2024. The Author(s).
Conflict of interest statement
F.T. consults for Immunai, Singularity Bio, CytoReason, Cellarity and Curie Bio Operations and has ownership interest in Dermagnostix and Cellarity. D.P. is on the scientific advisory board of Insitro. The other authors declare no competing interests.
Figures













References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
