

Synthetic surrogates improve power for genome-wide association studies of partially missing phenotypes in population biobanks
- PMID: 38872030
- PMCID: PMC11955959
- DOI: 10.1038/s41588-024-01793-9
Synthetic surrogates improve power for genome-wide association studies of partially missing phenotypes in population biobanks
Abstract
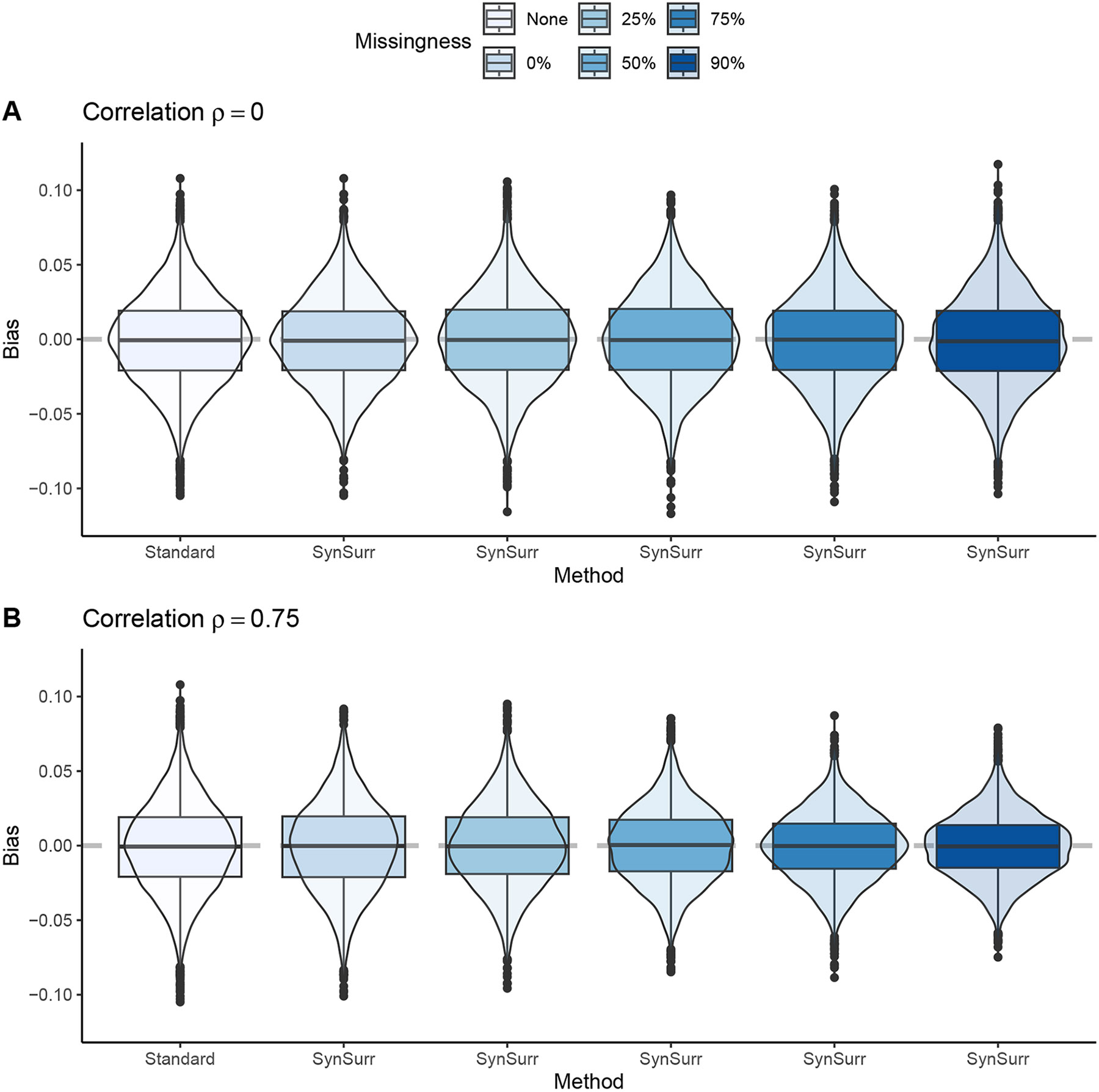
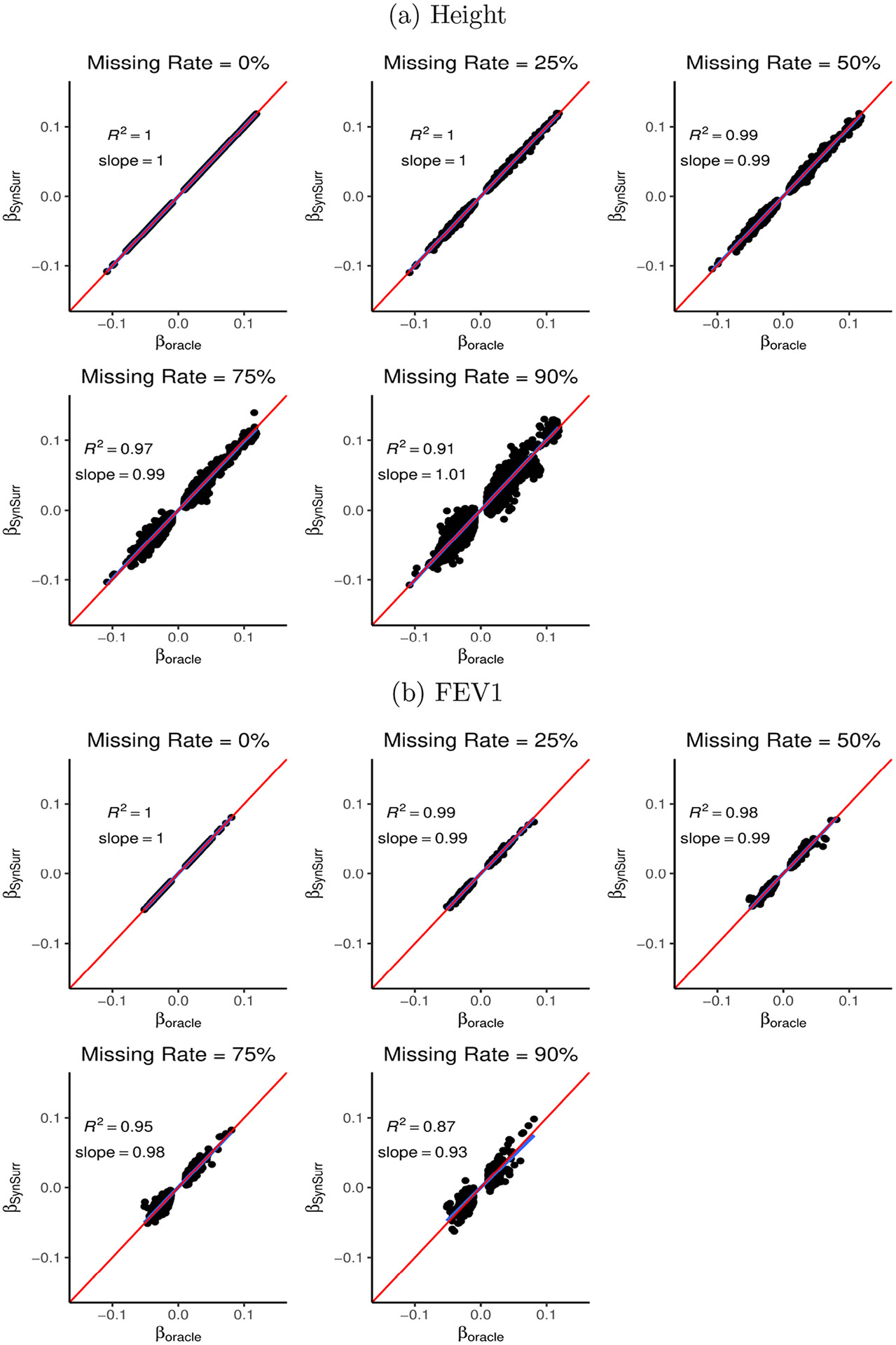
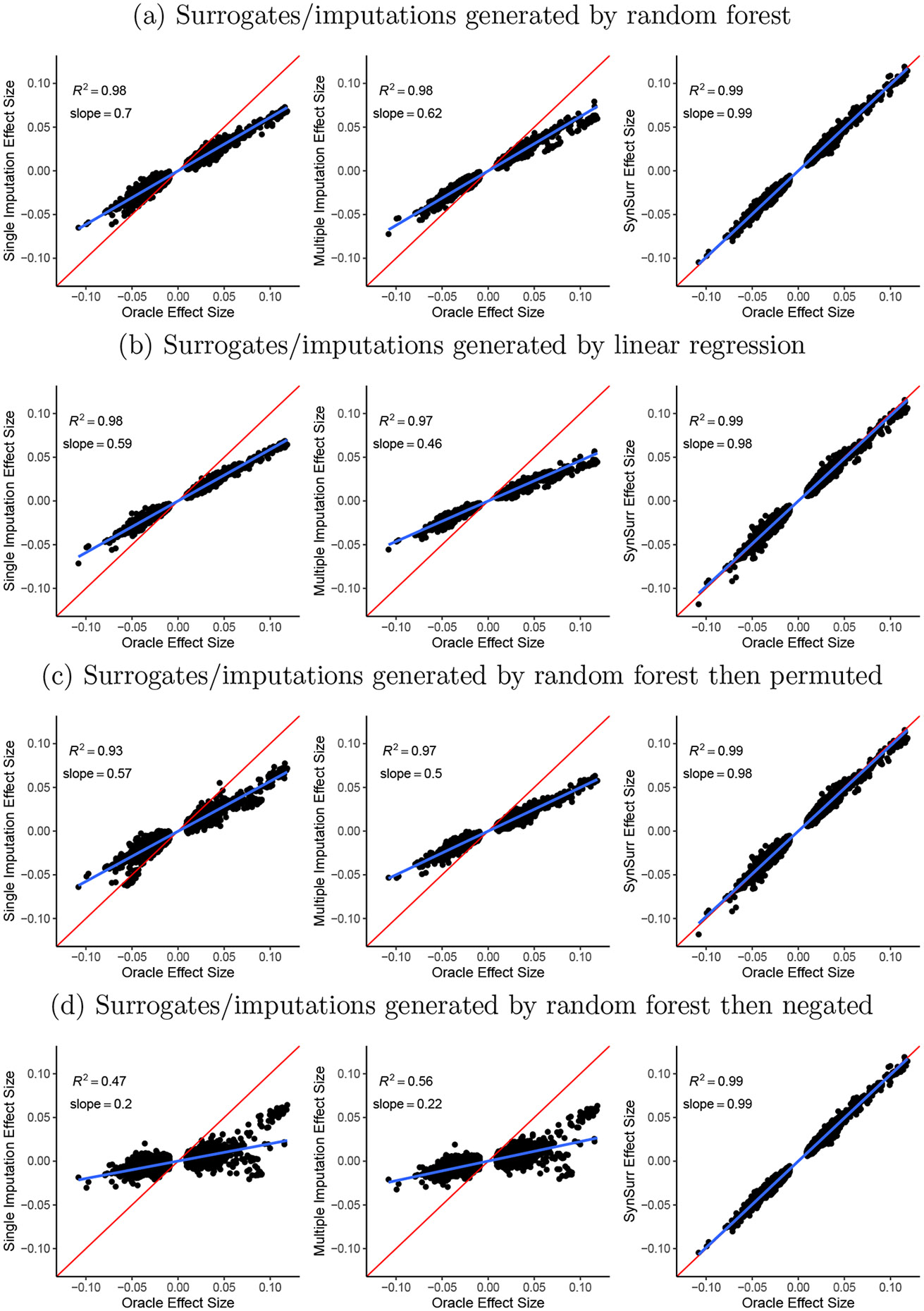
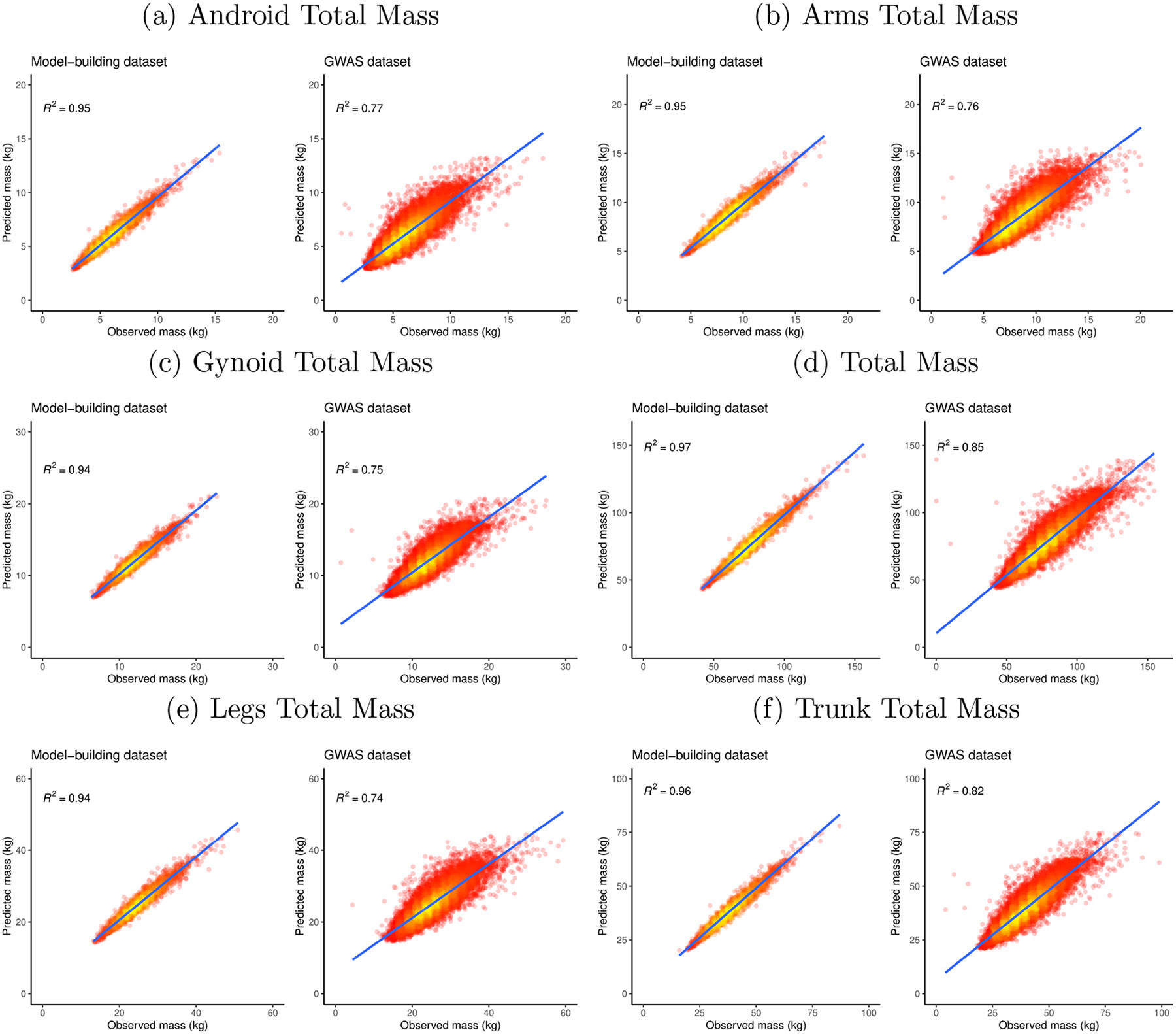
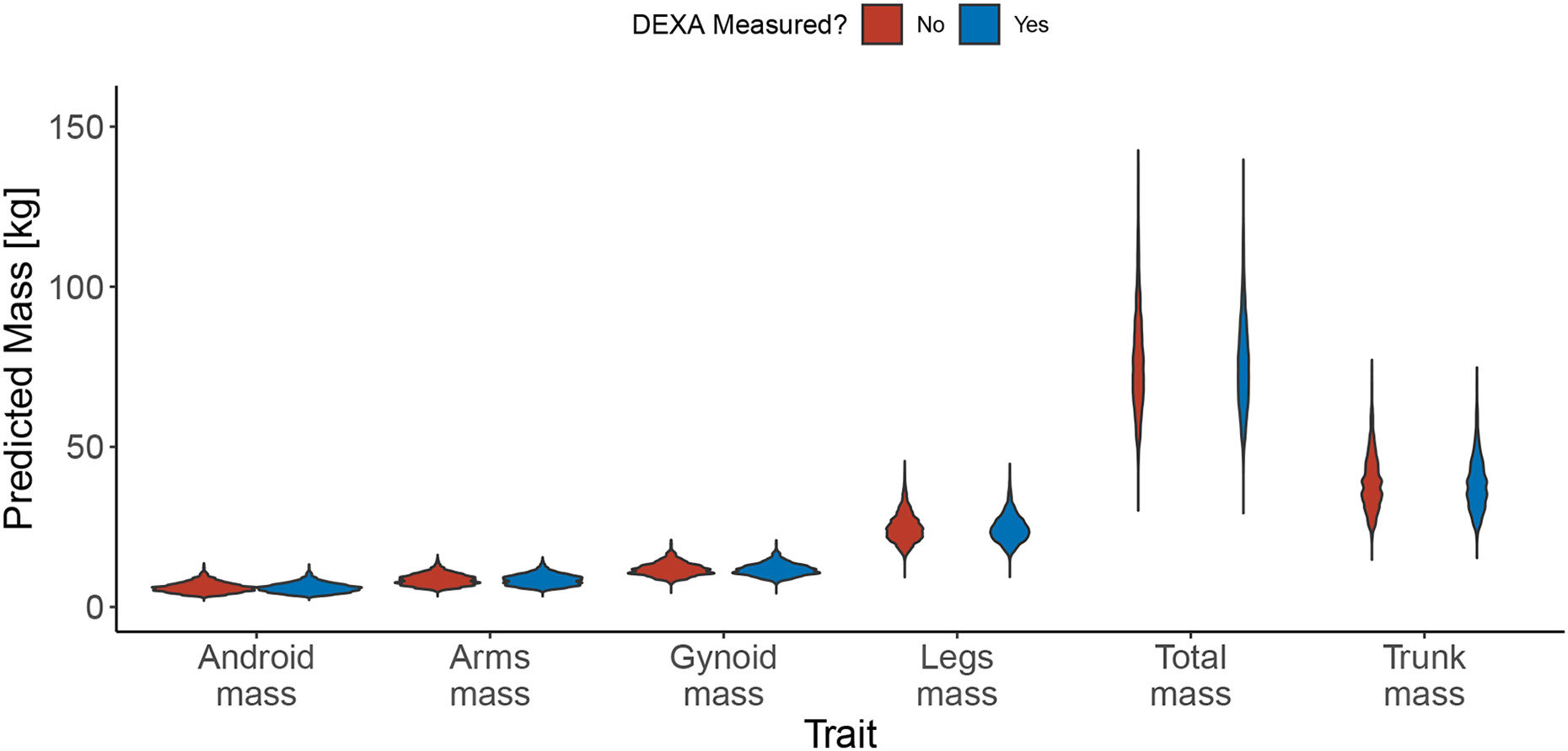
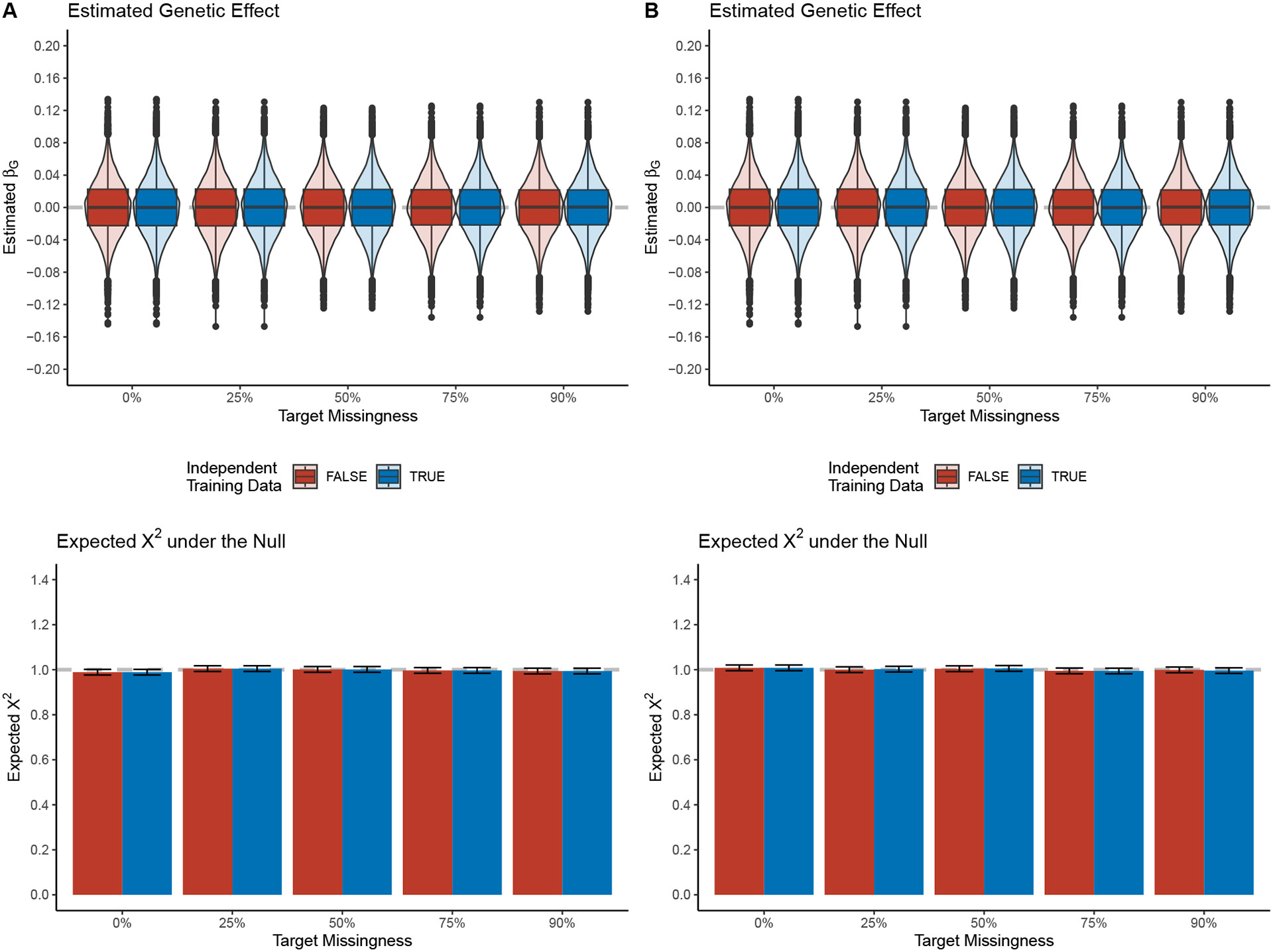
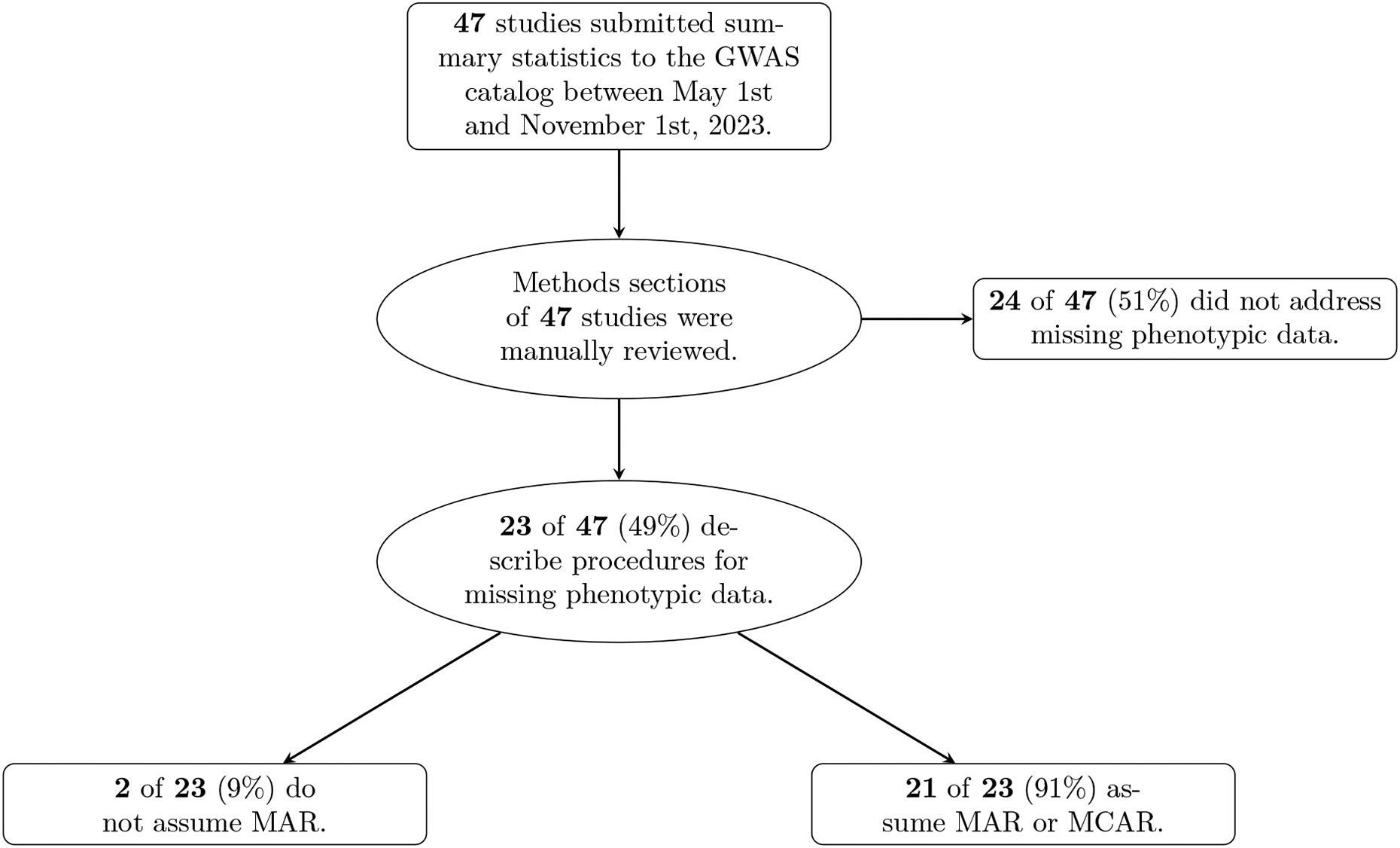
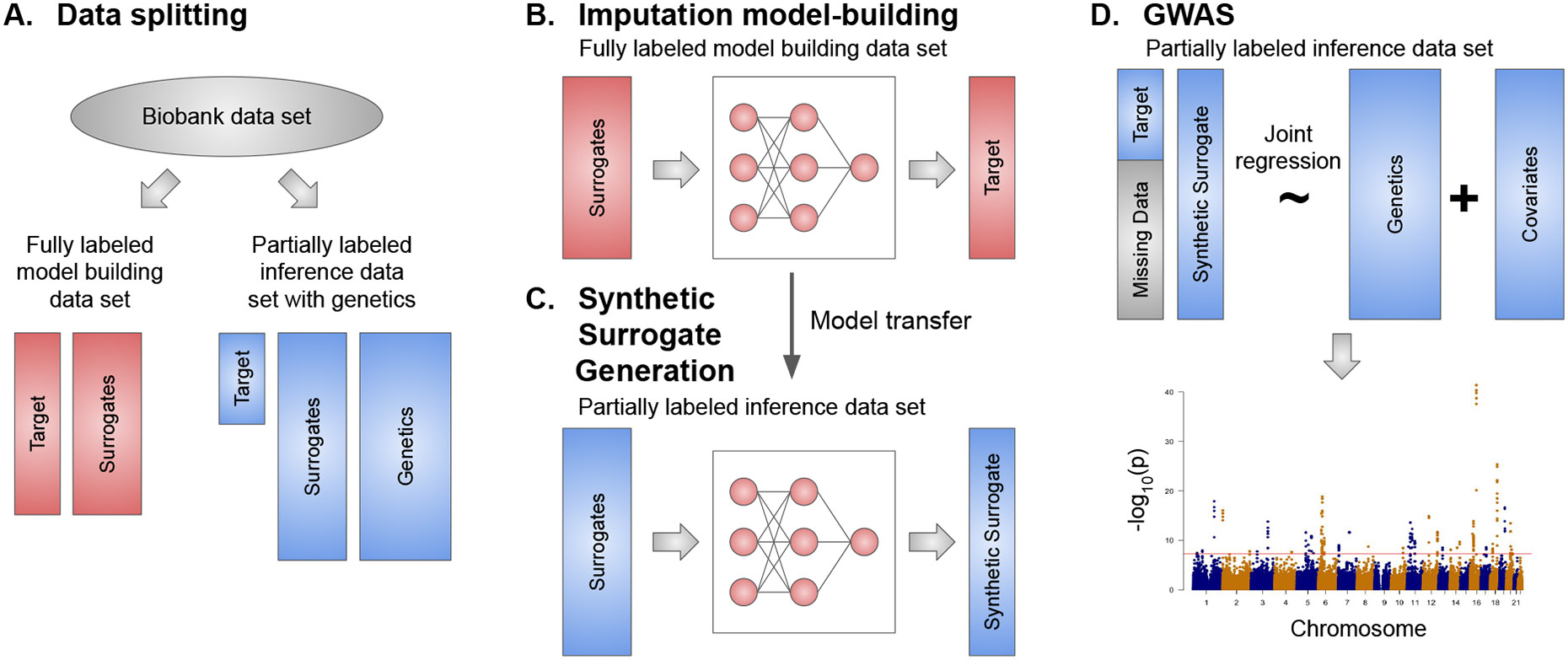
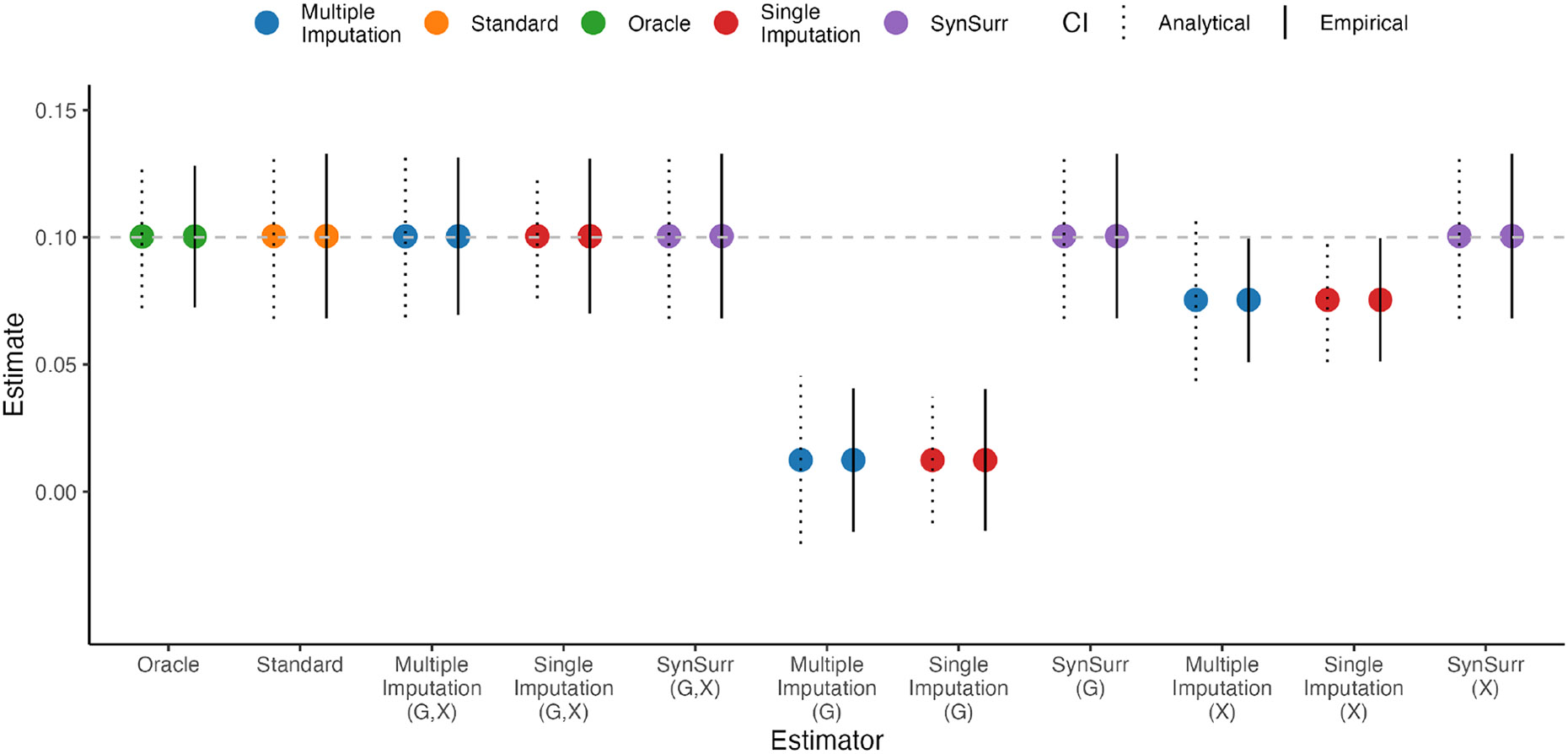
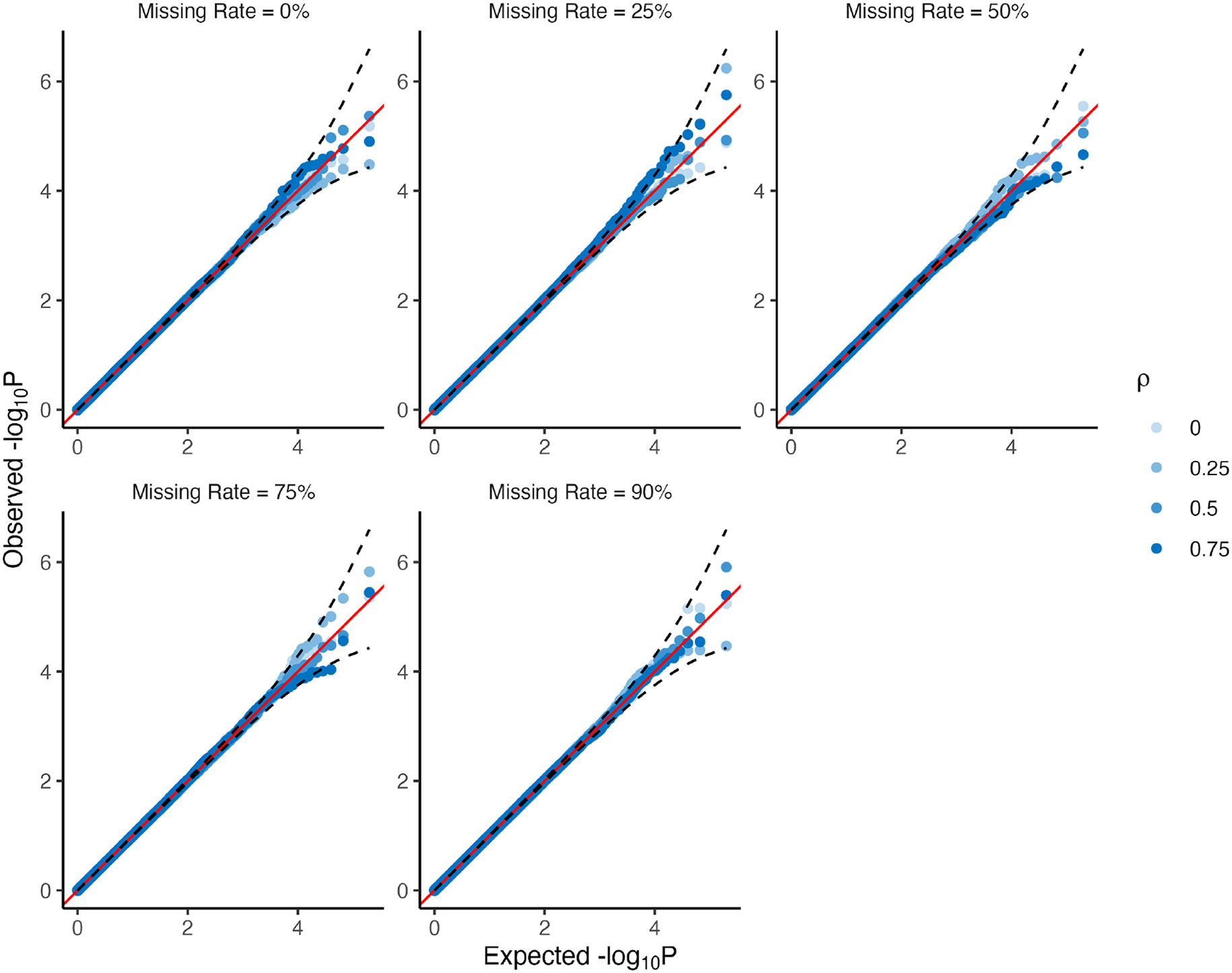
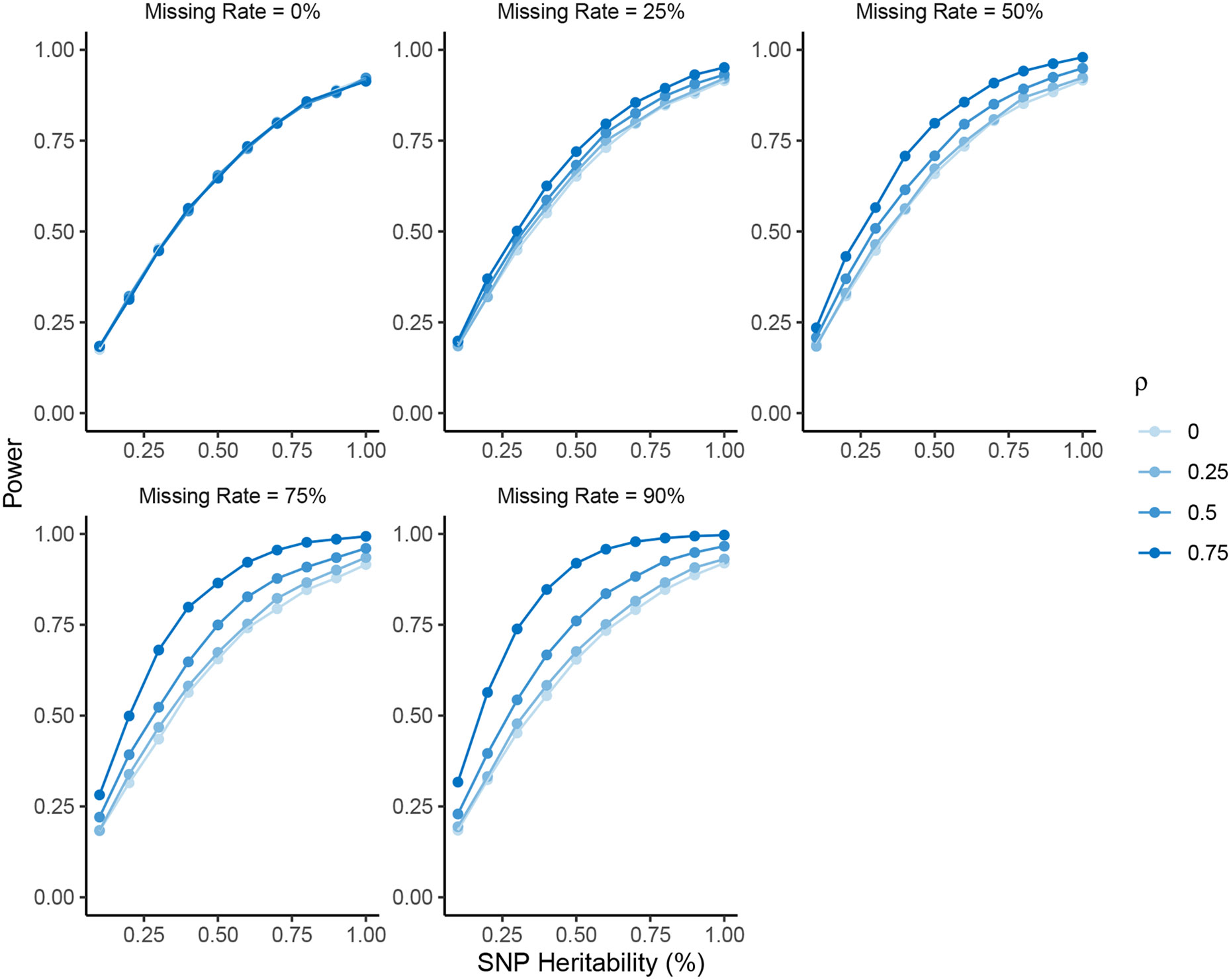
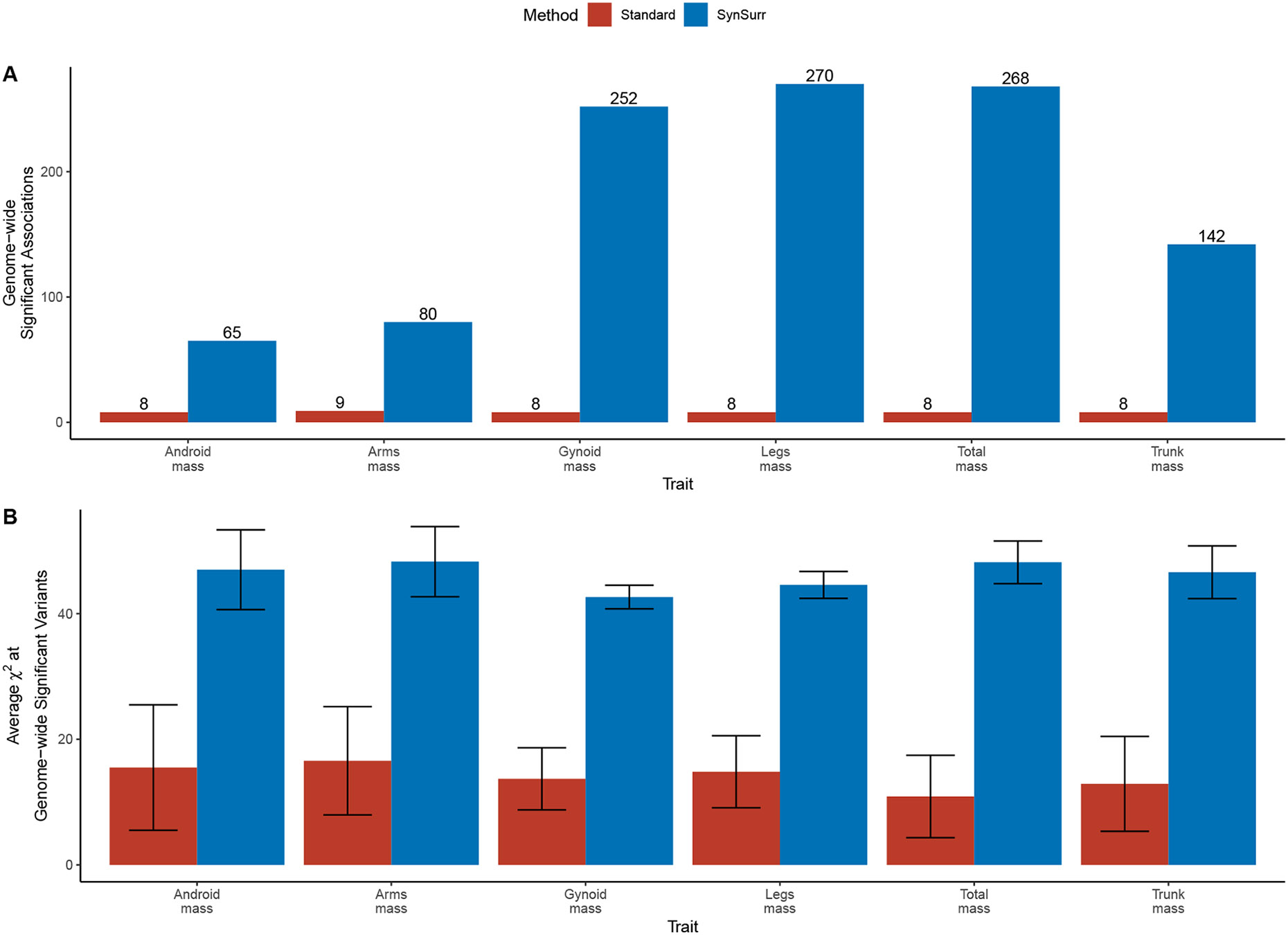
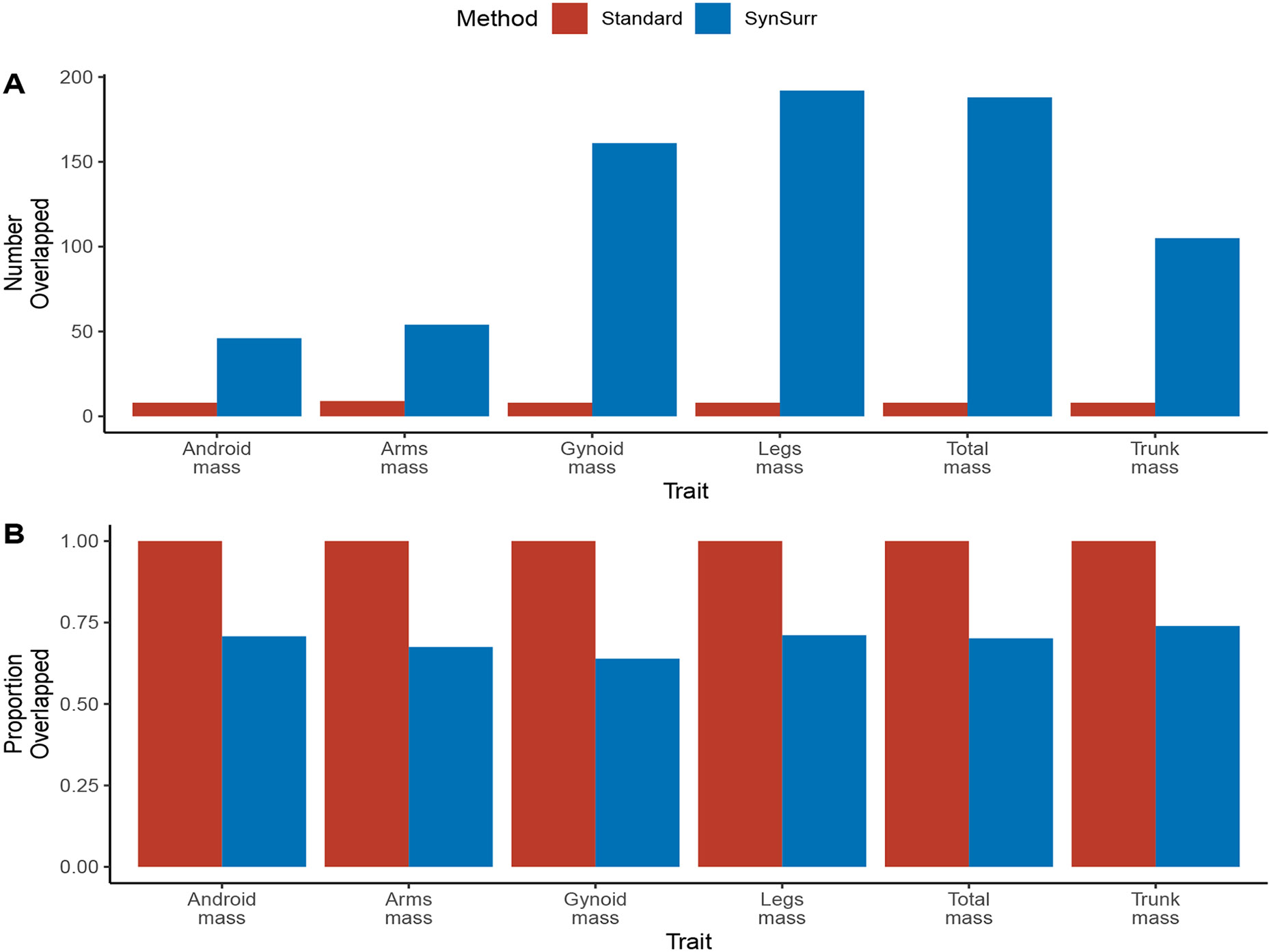
Within population biobanks, incomplete measurement of certain traits limits the power for genetic discovery. Machine learning is increasingly used to impute the missing values from the available data. However, performing genome-wide association studies (GWAS) on imputed traits can introduce spurious associations, identifying genetic variants that are not associated with the original trait. Here we introduce a new method, synthetic surrogate (SynSurr) analysis, which makes GWAS on imputed phenotypes robust to imputation errors. Rather than replacing missing values, SynSurr jointly analyzes the original and imputed traits. We show that SynSurr estimates the same genetic effect as standard GWAS and improves power in proportion to the quality of the imputations. SynSurr requires a commonly made missing-at-random assumption but relaxes the requirements of existing imputation methods by not requiring correct model specification. We present extensive simulations and ablation analyses to validate SynSurr and apply it to empower the GWAS of dual-energy X-ray absorptiometry traits within the UK Biobank.
© 2024. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
Similar articles
-
Valid inference for machine learning-assisted genome-wide association studies.Nat Genet. 2024 Nov;56(11):2361-2369. doi: 10.1038/s41588-024-01934-0. Epub 2024 Sep 30. Nat Genet. 2024. PMID: 39349818 Free PMC article.
-
Deep learning-based phenotype imputation on population-scale biobank data increases genetic discoveries.Nat Genet. 2023 Dec;55(12):2269-2276. doi: 10.1038/s41588-023-01558-w. Epub 2023 Nov 20. Nat Genet. 2023. PMID: 37985819 Free PMC article.
-
Trait imputation enhances nonlinear genetic prediction for some traits.Genetics. 2024 Nov 6;228(3):iyae148. doi: 10.1093/genetics/iyae148. Genetics. 2024. PMID: 39255064 Free PMC article.
-
Genotype Imputation from Large Reference Panels.Annu Rev Genomics Hum Genet. 2018 Aug 31;19:73-96. doi: 10.1146/annurev-genom-083117-021602. Epub 2018 May 23. Annu Rev Genomics Hum Genet. 2018. PMID: 29799802 Review.
-
The UK Biobank: A Shining Example of Genome-Wide Association Study Science with the Power to Detect the Murky Complications of Real-World Epidemiology.Annu Rev Genomics Hum Genet. 2022 Aug 31;23:569-589. doi: 10.1146/annurev-genom-121321-093606. Epub 2022 May 4. Annu Rev Genomics Hum Genet. 2022. PMID: 35508184 Review.
Cited by
-
Influence of multi-species data on gene-disease associations in substance use disorder using random walk with restart models.PLoS One. 2025 Jun 16;20(6):e0325201. doi: 10.1371/journal.pone.0325201. eCollection 2025. PLoS One. 2025. PMID: 40522980 Free PMC article.
-
A statistical framework for powerful multi-trait rare variant analysis in large-scale whole-genome sequencing studies.bioRxiv [Preprint]. 2023 Nov 2:2023.10.30.564764. doi: 10.1101/2023.10.30.564764. bioRxiv. 2023. Update in: Nat Comput Sci. 2025 Feb;5(2):125-143. doi: 10.1038/s43588-024-00764-8. PMID: 37961350 Free PMC article. Updated. Preprint.
-
A statistical framework for multi-trait rare variant analysis in large-scale whole-genome sequencing studies.Nat Comput Sci. 2025 Feb;5(2):125-143. doi: 10.1038/s43588-024-00764-8. Epub 2025 Feb 7. Nat Comput Sci. 2025. PMID: 39920506 Free PMC article.
-
Genetic association studies using disease liabilities from deep neural networks.Am J Hum Genet. 2025 Mar 6;112(3):675-692. doi: 10.1016/j.ajhg.2025.01.019. Epub 2025 Feb 21. Am J Hum Genet. 2025. PMID: 39986278 Free PMC article.
-
Valid inference for machine learning-assisted genome-wide association studies.Nat Genet. 2024 Nov;56(11):2361-2369. doi: 10.1038/s41588-024-01934-0. Epub 2024 Sep 30. Nat Genet. 2024. PMID: 39349818 Free PMC article.
References
-
- Gaziano JM et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. Journal of clinical epidemiology 70, 214–223 (2016). - PubMed
-
- Tan VY & Timpson NJ The UK Biobank: A Shining Example of Genome-Wide Association Study Science with the Power to Detect the Murky Complications of Real-World Epidemiology. Annual Review of Genomics and Human Genetics 23 (2022). - PubMed
Methods-only References
-
- Lawlor D, Harbord R, Sterne J, Timpson N & Smith G Mendelian randomization: Using genes as instruments for makingcausal inferences in epidemiology. Statistics in Medicine 27, 1133–1163 (2008). - PubMed
-
- McCaw Z. Surrogate Regression 10.5281/zenodo.10897842. - DOI
-
- Gao J, Gronsbell J & McCaw Z Synthetic Surrogate Analysis 10.5281/zenodo.10901237. - DOI
-
- McCaw ZR SurrogateRegression: Surrogate Outcome Regression Analysis Comprehensive R Archive Network (2020). https://CRAN.R-project.org/package=SurrogateRegression.
-
- Meng X-L & Rubin DB Maximum likelihood estimation via the ECM algorithm: A general framework. Biometrika 80, 267–278 (1993).
MeSH terms
Grants and funding
- U19-CA203654/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R35 CA197449/CA/NCI NIH HHS/United States
- U19 CA203654/CA/NCI NIH HHS/United States
- U01 HG012064/HG/NHGRI NIH HHS/United States
- R01 HL163560/HL/NHLBI NIH HHS/United States
- U01-HG012064/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- U01 HG009088/HG/NHGRI NIH HHS/United States
- RGPIN-2021-03734/Canadian Network for Research and Innovation in Machining Technology, Natural Sciences and Engineering Research Council of Canada (NSERC Canadian Network for Research and Innovation in Machining Technology)
- R35-CA197449/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- U01-HG009088/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01-HL163560/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- F31-HL140822/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- F31 HL140822/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
