

In silico screening of LRRK2 WDR domain inhibitors using deep docking and free energy simulations
- PMID: 38873063
- PMCID: PMC11168082
- DOI: 10.1039/d3sc06880c
In silico screening of LRRK2 WDR domain inhibitors using deep docking and free energy simulations
Abstract
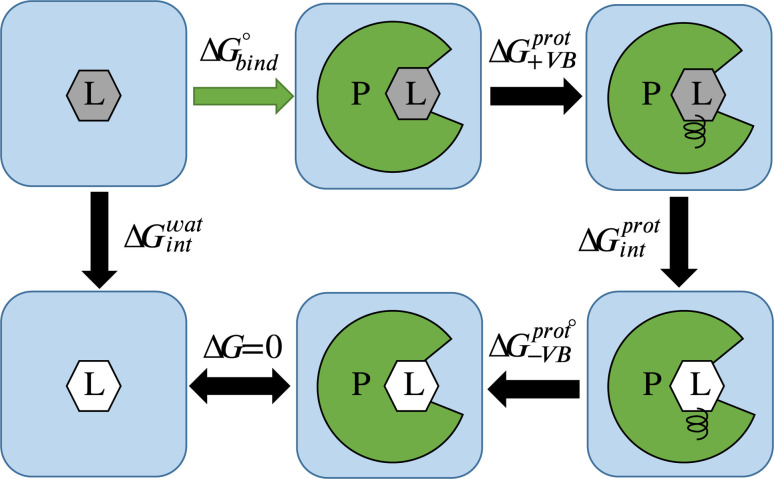
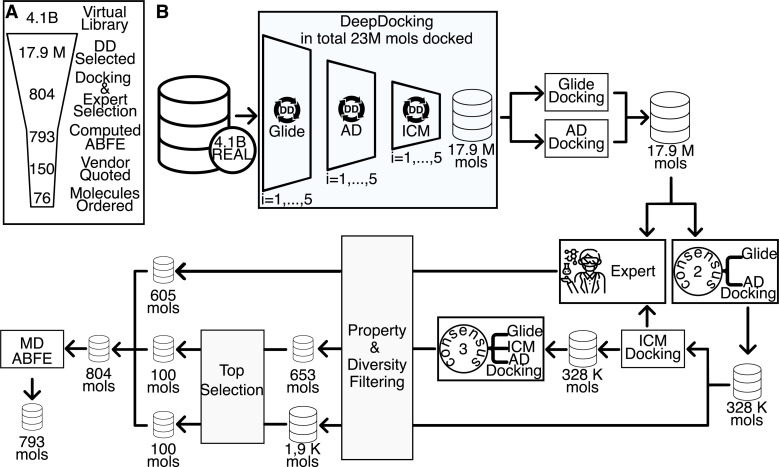
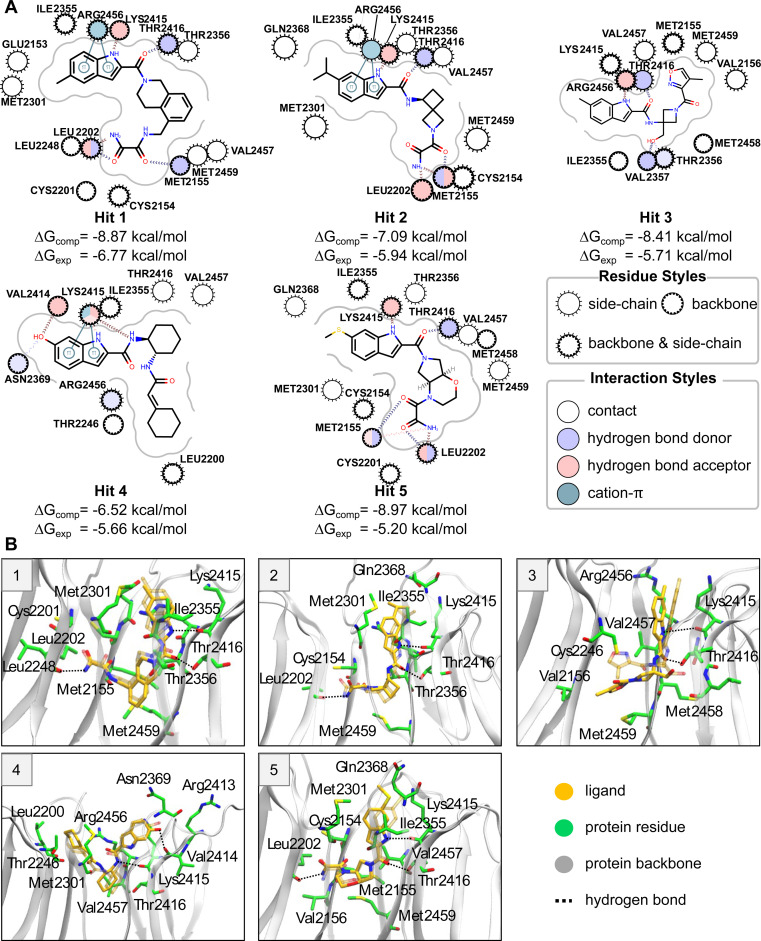
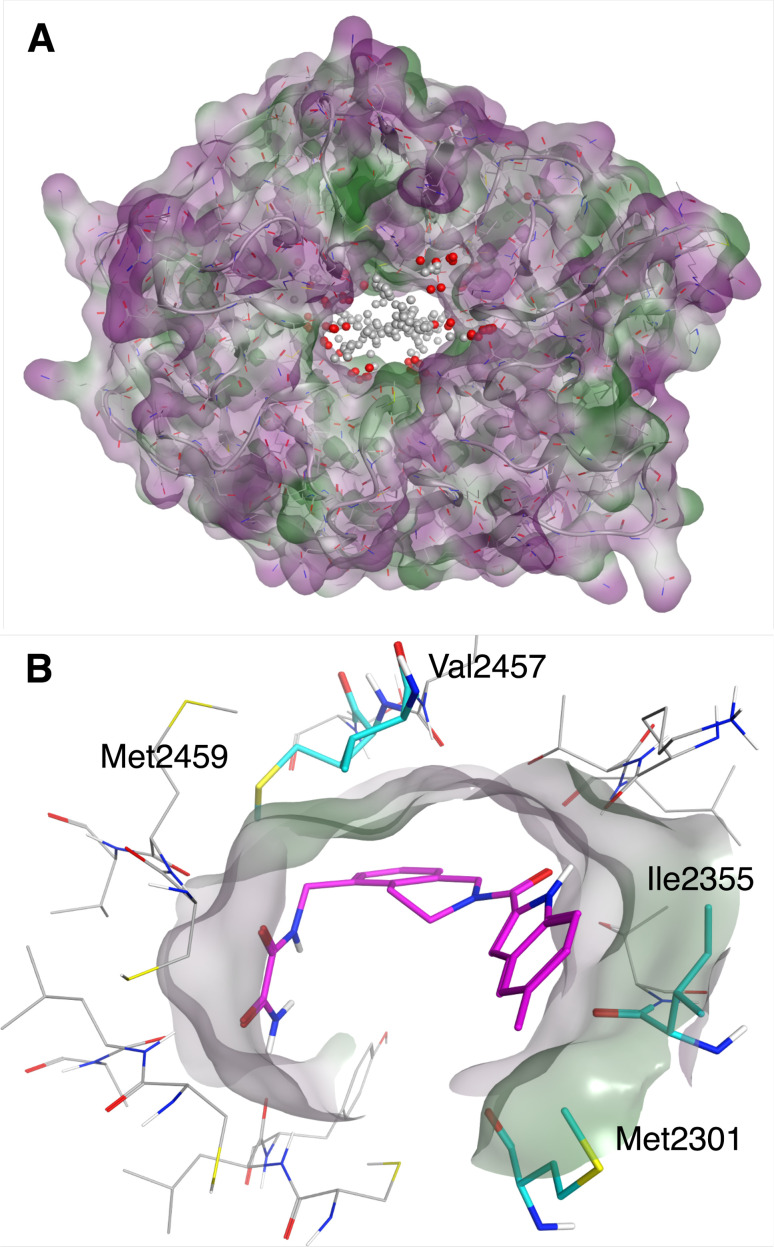
The Critical Assessment of Computational Hit-Finding Experiments (CACHE) Challenge series is focused on identifying small molecule inhibitors of protein targets using computational methods. Each challenge contains two phases, hit-finding and follow-up optimization, each of which is followed by experimental validation of the computational predictions. For the CACHE Challenge #1, the Leucine-Rich Repeat Kinase 2 (LRRK2) WD40 Repeat (WDR) domain was selected as the target for in silico hit-finding and optimization. Mutations in LRRK2 are the most common genetic cause of the familial form of Parkinson's disease. The LRRK2 WDR domain is an understudied drug target with no known molecular inhibitors. Herein we detail the first phase of our winning submission to the CACHE Challenge #1. We developed a framework for the high-throughput structure-based virtual screening of a chemically diverse small molecule space. Hit identification was performed using the large-scale Deep Docking (DD) protocol followed by absolute binding free energy (ABFE) simulations. ABFEs were computed using an automated molecular dynamics (MD)-based thermodynamic integration (TI) approach. 4.1 billion ligands from Enamine REAL were screened with DD followed by ABFEs computed by MD TI for 793 ligands. 76 ligands were prioritized for experimental validation, with 59 compounds successfully synthesized and 5 compounds identified as hits, yielding a 8.5% hit rate. Our results demonstrate the efficacy of the combined DD and ABFE approaches for hit identification for a target with no previously known hits. This approach is widely applicable for the efficient screening of ultra-large chemical libraries as well as rigorous protein-ligand binding affinity estimation leveraging modern computational resources.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures
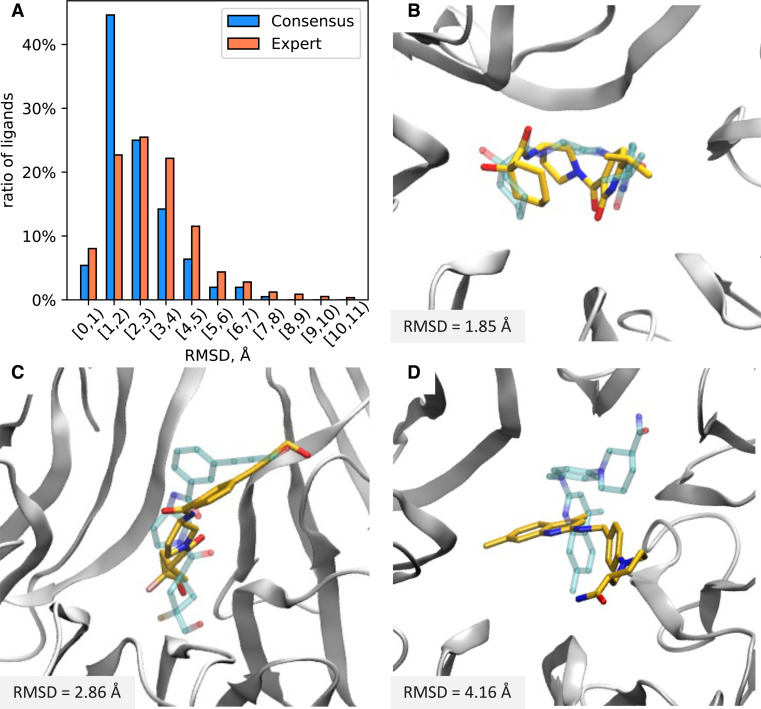
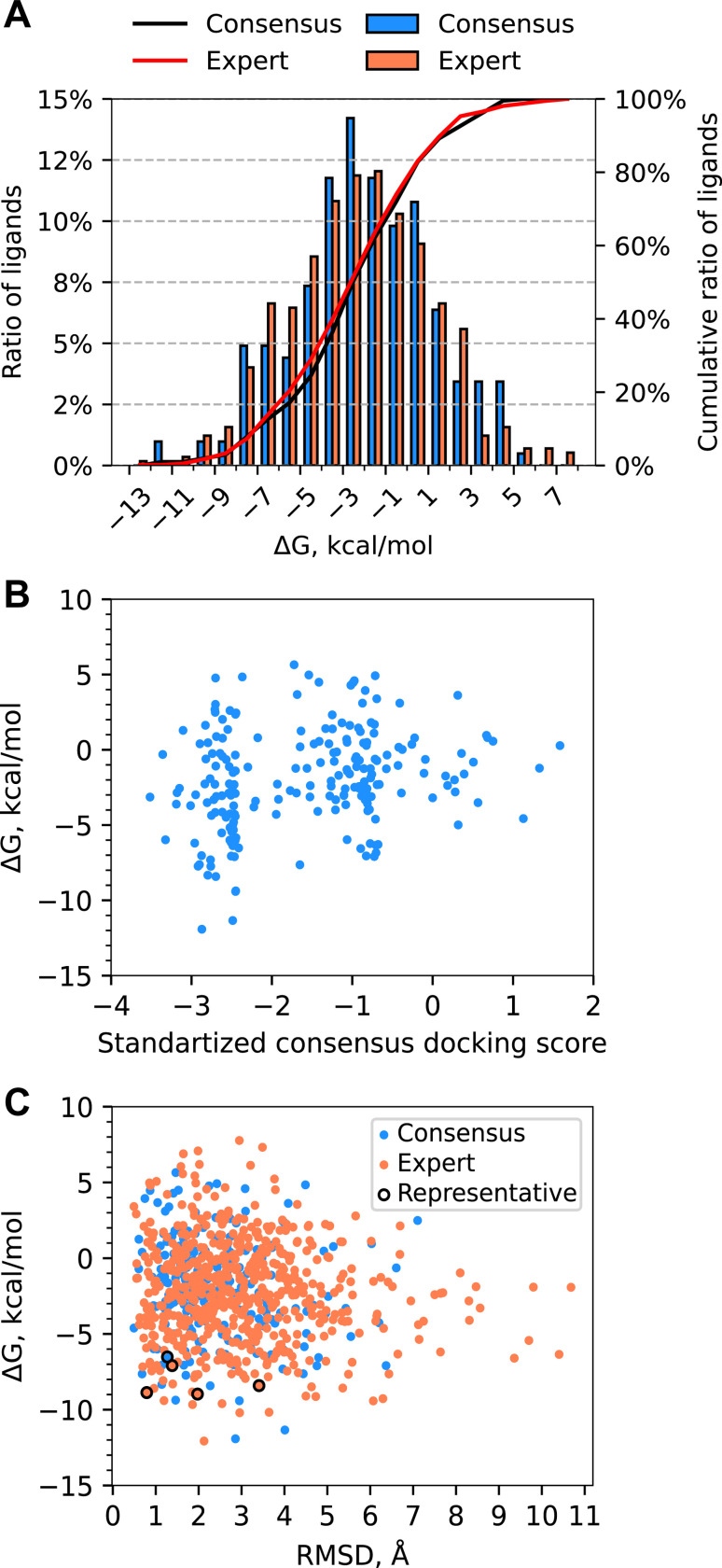
References
-
- Ackloo S. Al-Awar R. Amaro R. E. Arrowsmith C. H. Azevedo H. Batey R. A. et al., CACHE (Critical Assessment of Computational Hit-finding Experiments): a public-private partnership benchmarking initiative to enable the development of computational methods for hit-finding. Nat. Rev. Chem. 2022;6(4):287–295. doi: 10.1038/s41570-022-00363-z. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources