

Identification of a novel likely pathogenic TPM1 variant linked to hypertrophic cardiomyopathy in a family with sudden cardiac death
- PMID: 38874371
- PMCID: PMC11424302
- DOI: 10.1002/ehf2.14906
Identification of a novel likely pathogenic TPM1 variant linked to hypertrophic cardiomyopathy in a family with sudden cardiac death
Abstract
Aims: Hypertrophic cardiomyopathy (HCM) is an autosomal dominant genetic cardiac disorder characterized by unexplained left ventricular hypertrophy. It can cause a wide spectrum of clinical manifestations, ranging from asymptomatic to heart failure and sudden cardiac death (SCD). Approximately half of HCM cases are caused by variants in sarcomeric proteins, including α-tropomyosin (TPM1). In this study, we aimed to characterize the clinical and molecular phenotype of HCM in an Iranian pedigree with SCD.
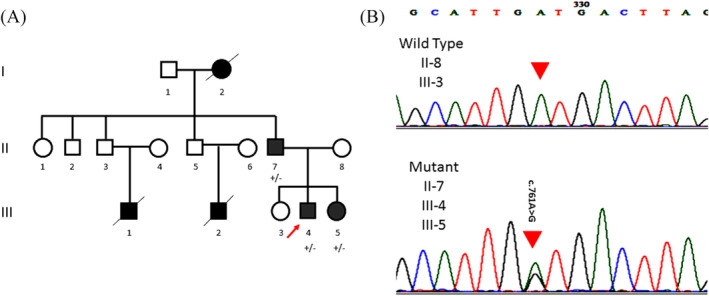
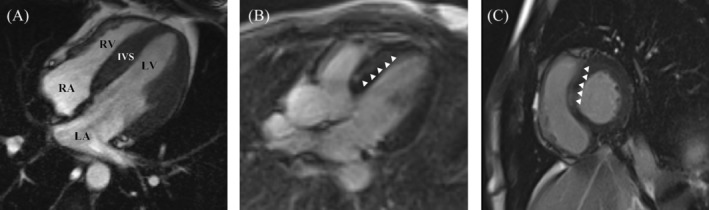
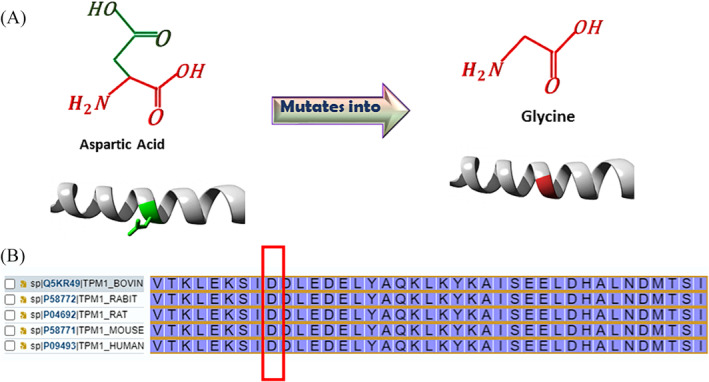
Methods and results: The proband and available family members underwent comprehensive clinical evaluations, including echocardiography, cardiac magnetic resonance (CMR) imaging and electrocardiography (ECG). Whole-exome sequencing (WES) was performed in all available family members to identify the causal variant, which was validated, and segregation analysis was conducted via Sanger sequencing. WES identified a novel missense variant, c.761A>G:p.D254G (NM_001018005.2), in the TPM1 gene, in the proband, his father and one of his sisters. Bioinformatic analysis predicted it to be likely pathogenic. Clinical features in affected individuals were consistent with HCM.
Conclusions: The identification of a novel TPM1 variant in a family with HCM and SCD underscores the critical role of genetic screening in at-risk families. Early detection of pathogenic variants can facilitate timely intervention and management, potentially reducing the risk of SCD in individuals with HCM.
Keywords: genetic; hypertrophic cardiomyopathy; tropomyosin; variant; whole‐exome sequencing.
© 2024 The Author(s). ESC Heart Failure published by John Wiley & Sons Ltd on behalf of European Society of Cardiology.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
Similar articles
-
Hypertrophic cardiomyopathy caused by a novel alpha-tropomyosin mutation (V95A) is associated with mild cardiac phenotype, abnormal calcium binding to troponin, abnormal myosin cycling, and poor prognosis.Circulation. 2001 Jan 2;103(1):65-71. doi: 10.1161/01.cir.103.1.65. Circulation. 2001. PMID: 11136687
-
Inducibility of life-threatening ventricular arrhythmias is related to maximum left ventricular thickness and clinical markers of sudden cardiac death in patients with hypertrophic cardiomyopathy attributable to the Asp175Asn mutation in the alpha-tropomyosin gene.J Mol Cell Cardiol. 2004 Jan;36(1):91-9. doi: 10.1016/j.yjmcc.2003.10.003. J Mol Cell Cardiol. 2004. PMID: 14734051
-
A novel homozygous TPM1 mutation in familial pediatric hypertrophic cardiomyopathy and in silico screening of potential targeting drugs.Eur Rev Med Pharmacol Sci. 2020 Jul;24(14):7732-7744. doi: 10.26355/eurrev_202007_22299. Eur Rev Med Pharmacol Sci. 2020. PMID: 32744700
-
Natural history and clinical outcomes of patients with hypertrophic cardiomyopathy from thin filament mutations.ESC Heart Fail. 2024 Dec;11(6):3501-3510. doi: 10.1002/ehf2.14848. Epub 2024 May 21. ESC Heart Fail. 2024. PMID: 38773858 Free PMC article. Review.
-
Sudden cardiac death in patients with hypertrophic cardiomyopathy: from bench to bedside with an emphasis on genetic markers.Clin Cardiol. 1995 Apr;18(4):189-98. doi: 10.1002/clc.4960180403. Clin Cardiol. 1995. PMID: 7788945 Review.
Cited by
-
Protein Sequencing with Single Amino Acid Resolution Discerns Peptides That Discriminate Tropomyosin Proteoforms.J Proteome Res. 2025 Aug 1;24(8):3798-3807. doi: 10.1021/acs.jproteome.4c00978. Epub 2025 Jun 10. J Proteome Res. 2025. PMID: 40493858 Free PMC article.
References
-
- García‐Vielma C, Lazalde‐Córdova LG, Arzola‐Hernández JC, González‐Aceves EN, López‐Zertuche H, Guzmán‐Delgado NE, et al. Identification of variants in genes associated with hypertrophic cardiomyopathy in Mexican patients. Mol Genet Genomics [Internet] 2023;298:1289‐1299. doi:10.1007/s00438-023-02048-8 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
