

Biases in ARG-Based Inference of Historical Population Size in Populations Experiencing Selection
- PMID: 38874402
- PMCID: PMC11245712
- DOI: 10.1093/molbev/msae118
Biases in ARG-Based Inference of Historical Population Size in Populations Experiencing Selection
Abstract
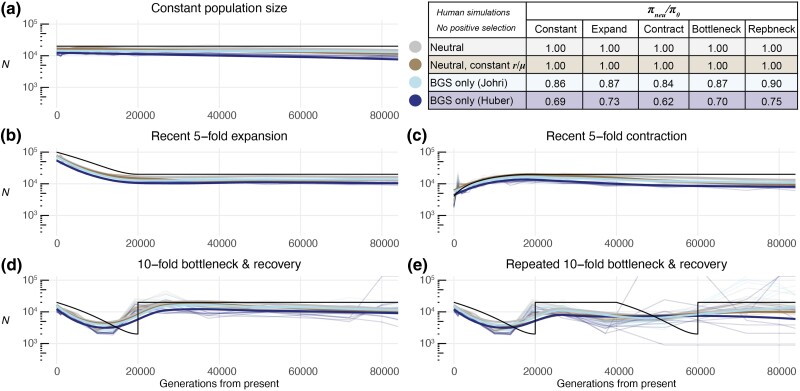
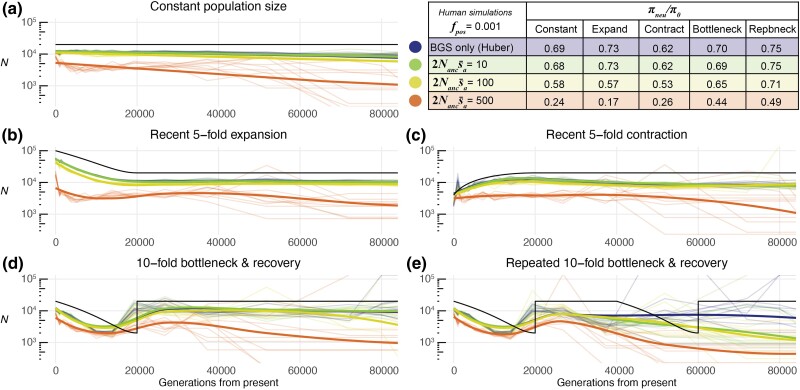
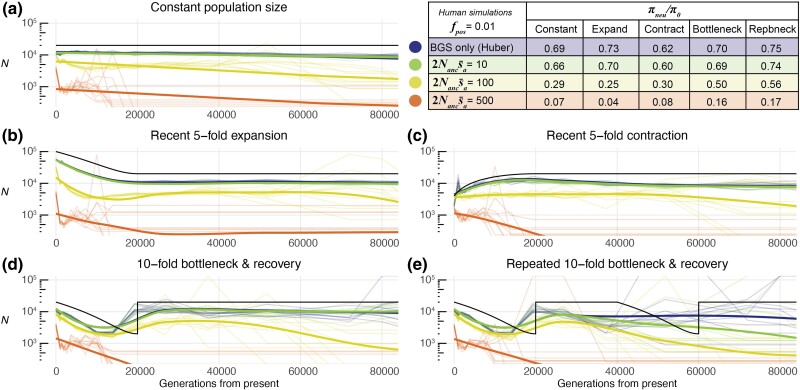
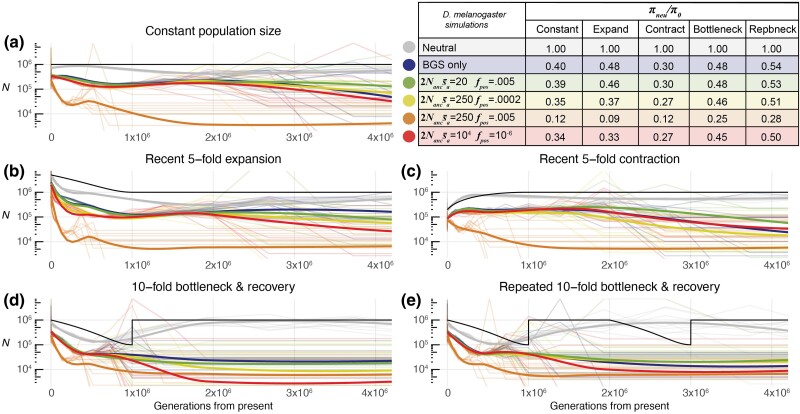
Inferring the demographic history of populations provides fundamental insights into species dynamics and is essential for developing a null model to accurately study selective processes. However, background selection and selective sweeps can produce genomic signatures at linked sites that mimic or mask signals associated with historical population size change. While the theoretical biases introduced by the linked effects of selection have been well established, it is unclear whether ancestral recombination graph (ARG)-based approaches to demographic inference in typical empirical analyses are susceptible to misinference due to these effects. To address this, we developed highly realistic forward simulations of human and Drosophila melanogaster populations, including empirically estimated variability of gene density, mutation rates, recombination rates, purifying, and positive selection, across different historical demographic scenarios, to broadly assess the impact of selection on demographic inference using a genealogy-based approach. Our results indicate that the linked effects of selection minimally impact demographic inference for human populations, although it could cause misinference in populations with similar genome architecture and population parameters experiencing more frequent recurrent sweeps. We found that accurate demographic inference of D. melanogaster populations by ARG-based methods is compromised by the presence of pervasive background selection alone, leading to spurious inferences of recent population expansion, which may be further worsened by recurrent sweeps, depending on the proportion and strength of beneficial mutations. Caution and additional testing with species-specific simulations are needed when inferring population history with non-human populations using ARG-based approaches to avoid misinference due to the linked effects of selection.
Keywords: Drosophila melanogaster; ancestral recombination graph; background selection; demographic inference; human population genomics; selective sweeps.
© The Author(s) 2024. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Conflict of interest statement
Conflict of Interest The authors declare no conflicts of interest.
Figures
References
-
- Beichman AC, Huerta-Sanchez E, Lohmueller KE. Using genomic data to infer historic population dynamics of nonmodel organisms. Annu Rev Ecol Evol Syst. 2018:49(1):433–456. 10.1146/annurev-ecolsys-110617-062431. - DOI
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
