

The polyglutamine protein ATXN2: from its molecular functions to its involvement in disease
- PMID: 38877004
- PMCID: PMC11178924
- DOI: 10.1038/s41419-024-06812-5
The polyglutamine protein ATXN2: from its molecular functions to its involvement in disease
Abstract
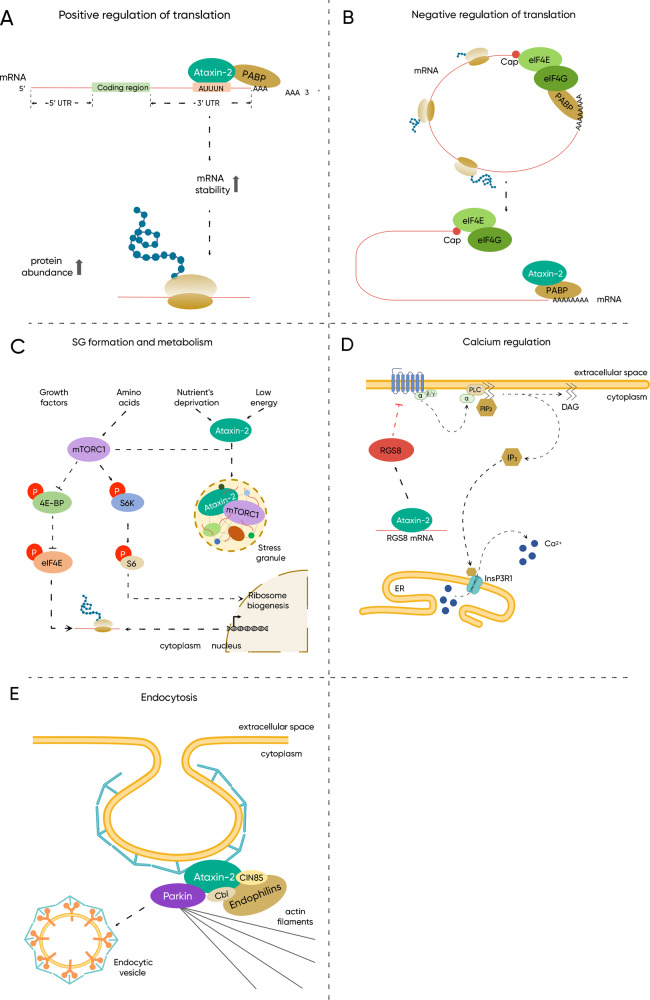
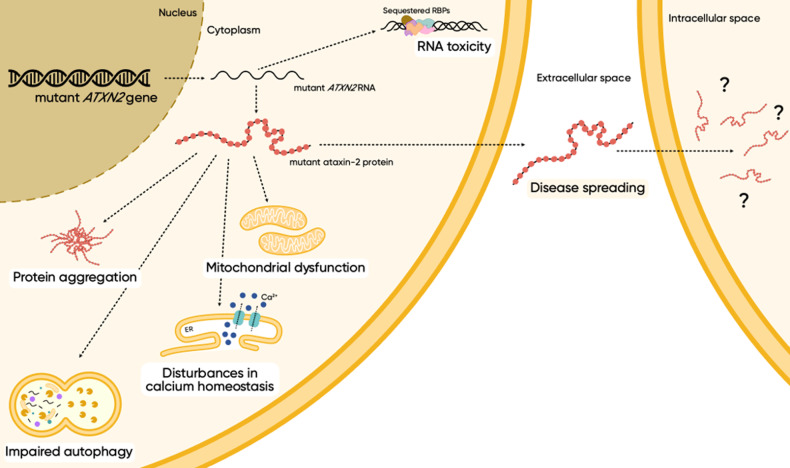
A CAG repeat sequence in the ATXN2 gene encodes a polyglutamine (polyQ) tract within the ataxin-2 (ATXN2) protein, showcasing a complex landscape of functions that have been progressively unveiled over recent decades. Despite significant progresses in the field, a comprehensive overview of the mechanisms governed by ATXN2 remains elusive. This multifaceted protein emerges as a key player in RNA metabolism, stress granules dynamics, endocytosis, calcium signaling, and the regulation of the circadian rhythm. The CAG overexpansion within the ATXN2 gene produces a protein with an extended poly(Q) tract, inducing consequential alterations in conformational dynamics which confer a toxic gain and/or partial loss of function. Although overexpanded ATXN2 is predominantly linked to spinocerebellar ataxia type 2 (SCA2), intermediate expansions are also implicated in amyotrophic lateral sclerosis (ALS) and parkinsonism. While the molecular intricacies await full elucidation, SCA2 presents ATXN2-associated pathological features, encompassing autophagy impairment, RNA-mediated toxicity, heightened oxidative stress, and disruption of calcium homeostasis. Presently, SCA2 remains incurable, with patients reliant on symptomatic and supportive treatments. In the pursuit of therapeutic solutions, various studies have explored avenues ranging from pharmacological drugs to advanced therapies, including cell or gene-based approaches. These endeavours aim to address the root causes or counteract distinct pathological features of SCA2. This review is intended to provide an updated compendium of ATXN2 functions, delineate the associated pathological mechanisms, and present current perspectives on the development of innovative therapeutic strategies.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
